 | |||||||||
|  | ||||||||
| |||||||||
| | ||||||||
| kategória | ||||||||||
|
|
||||||||||
|
|
||
REAKCIÓKINETIKA
A reakciókinetika tárgya és a reakciók osztályozása
A termodinamikailag lehetséges reakciók a hömérséklettöl, a nyomástól, a reagáló anyag mennyiségétöl stb. függöen a legkülönbözöbb sebességgel, utakon és mechanizmus szerint játszódhatnak le. A kémiai átalakuláshoz idö szükséges, amely tág határok között változik, hiszen vannak nagyon gyorsan, pillanatszerüen lejátszódó reakciók, és vannak igen lassan végbemenö reakciók is. A reakciókinetika a kémiai folyamatok sebességét meghatározó törvényszerüségekkel és a reakciómechanizmus kiderítésével foglalkozik.
A reakciókat osztályozhatjuk aszerint, hogy a kiindulási anyagok vagy termékek milyen fázisban vannak. A homogén reakciókban résztvevö valamennyi anyag ugyanabban a fázisban vesz részt, ezek közül legegyszerübbek a homogén gázreakciók. A heterogén reakcióknál a reakcióban résztvevö anyagok különbözö fázisban vannak, a reakció csak az érintkezö fázisok határfelületén játszódik le.
A termodinamikailag lehetséges reakciók egy idöintervallumban teljesen vagy részlegesen mehetnek végbe. A teljes reakció során a kiindulási anyagok teljesen átalakulnak végtermékké, a részlegesen végbemenö (egyensúlyra vezetö) reakciók leggyakoribb típusa pedig a megfordítható folyamat. Ekkor a termékek a termodinamikailag lehetséges reakcióban visszaalakulnak. Szigorúan nézve minden reakció megfordítható, de a teljes reakciók egyensúlya annyira a végtermék irányába van eltolva, hogy az egyensúly beállta után a kiindulási anyagok már nem mutathatók ki.
Reakciósebesség
A kémiai folyamatok sebessége függ a reagáló anyagok koncentrációjától és a hömérséklettöl. A reakciók idöbeni lefolyásáról információval szolgálhat minden olyan változás, mely egyértelmüen kapcsolatba hozható a kémiai reakcióval. Ilyen lehet pl. a gázok közötti reakcióknál a nyomás, oldatokban végbemenö reakcióknál pedig a koncentrációk megváltozása.
A reakciósebesség a reakciókinetika központi fogalma; az idöegység alatti koncentrációváltozást jelenti. Végtelenül kis változásokra felírva:
v dc / dt,
ahol a pozitív elöjel a termékre ill. a termékek egyikére,
a negatív elöjel pedig valamelyik kiindulási anyagra vonatkozik.
Elegendö a reakcióegyenletben
szereplö egyetlen anyag koncentrációjának változását figyelemmel kísérni,
mivel a sztöchiometriai arányszámok ismeretében a különbözö anyagokra
definiált reakciósebességek kiszámíthatók egymásból. A reakciósebesség SI
mértékegysége: mol.m-3.s-
Elemi reakciók, molekularitás
A sztöchiometriai egyenletek általában csak a bruttó folyamatokat tükrözik, és nem adnak felvilágosítást a molekuláris szinten lejátszódó, sokszor igen bonyolult történésekre. A legtöbb átalakulás összetett, azaz több egymás után és mellett végbemenö, egyszerü reakciókból áll. Az összetett reakciókat adó egyszerü folyamatokat elemi reakcióknak, vagy reakciólépéseknek nevezzük. Egy reakció mechanizmusát akkor ismerjük, ha megállapítjuk, hogy az milyen elemi reakciókból tevödik össze, és kiderítjük a reakciólépéseket meghatározó tényezöket. Így pl. a nitrogén-dioxid és a szén-monoxid
NO2(g) + CO(g) =NO(g) + CO2(g)
bruttó egyenlettel leírt reakciója valójában két elemi lépésböl áll, ha a hömérséklet nem haladja meg az 500 oC-ot:
NO2(g) + NO2(g) = NO3(g) + NO(g),
NO3(g) + CO(g) = NO2(g) + CO2(g).
A reakció molekularitása a reakció mechanizmusával összefüggö fogalom amely megmutatja, hogy az adott kémiai reakció hány molekula kémiai kölcsönhatása során jön létre. Molekularitásról csak egyszerü elemi reakcióknál ill. összetett reakciók elemi lépései esetén beszélhetünk. A leggyakoribb az, hogy két részecske ütközése révén ún. bimolekuláris reakció játszódik le. Példa erre pl. az ózon reakciója naszcensz oxigénnel, amely oxigénmolekulát eredményez:
O3(g) + O(g) = O2(g) + O2(g).
Bimolekuláris elemi reakciók az oldatokban is gyakoriak. Ilyen pl. a hidroxóniumion és a hidroxidion reakciója vizes fázisban, ill. a metil-bromid és a kloridion reakciója acetonban:
H3O+ + OH = 2H2O(l) (vizes fázisban),
CH3Br + Cl = CH3Cl + Br (acetonban).
Szintén elég gyakoriak az ún. monomolekuláris (unimolekulás) kémiai reakciók, melyek során a molekulák belsö instabilitásuk folytán bomlanak el vagy alakulnak át izomerekké. Ezekhez némiképp hasonló a radioaktív bomlás, amely azonban az atommagok instabilitásának következménye, s így nem sorolható a kémiai folyamatok közé. A monomolekulás reakciók tipikus példája az etilamin gázfázisban lejátszódó höbomlása, valamint az ammónium-izocianát izomerizációja karbamiddá höhatásra:
C2H5NH2(g) = C2H4(g) + NH3(g),
NH4OCN(s) = CO(NH2)2(s).
A monomolekuláris reakcióban ugyan egyetlen anyag alakul át, ennek ellenére az eredményes átalakulást több eredménytelen ütközés elözi meg, melynek során a molekula belsö energiája lényegesen megnö, majd ütközés nélkül átalakul. Az elemi lépésben valójában egyetlen molekula szerepel, de ennek aktiválásához eredménytelen ütközések sokasága szükséges.
Trimolekulás reakciók során három részecske ütközik egyidejüleg. Ennek valószínüsége igen csekély, ezért trimolekuláris reakciók csak igen ritkán fordulnak elö. A nitrogén-oxid nitrogén-dioxiddá történö átalakulása során 2 nitrogén-oxid molekula ütközik egy oxigénmolekulával:
2 NO(g) + O2(g) = 2 NO2(g).
A háromnál nagyobb molekularitású reakció valószínüsége gyakorlatilag nulla.
Reakciósebesség- és rendüség
A kémiai reakció sebessége az idöegység alatt átalakuló anyagmennyiséget adja meg. A sebességet a koncentrációra szokás vonatkoztatni. A reakcióban résztvevö anyagok koncentrációi a reakcióidö elörehaladtával változnak, ezért változik a reakciósebesség is. A reakciósebességet akár a kiindulási anyagok, akár a termékek koncentrációjának változására vonatkoztathatjuk, ezért egy A + B C reakcióban differenciális forma helyett újra differenciákat alkalmazva a sebességet
(v) többféleképpen is kifejezhetjük:
v = ![]() ,
,
ahol D[A] jelenti az egyik kiindulási anyag koncentráció változását Dt idö alatt.
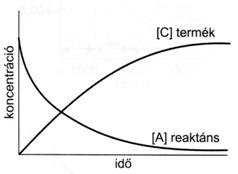
54. ábra A reaktáns és a termék koncentrációjának idöbeli változása (A C)
A reakciósebesség csak akkor jól meghatározott, ha tetszés szerinti kicsiny dt idö alatt bekövetkezett dc koncentrációváltozásra vonatkoztatjuk:
v =
![]()
A reakciósebesség kifejezésében a pozitív elöjelet a termékekre használjuk, mivel ezek koncentrációja nö a reakció során; a negatív elöjelet pedig a kiindulási anyagokra használjuk, mivel ezek koncentrációja csökken a reakció folyamán. A kémiai reakciók sebességét kísérletileg meg lehet határozni. A hidrogén-jodid képzödése során gáztérben a következö reakció játszódik le:
H2(g) + I2(g) = 2 HI(g).
Ahhoz, hogy a reakció lejátszódjon, a molekuláknak ütközni kell egymással. Az ütközések kis részét követi csak kémiai átalakulás, de a sikeres ütközések száma arányos az összes ütközések számával. A koncentráció, vagy a nyomás növelésével az ütközések száma is nö, ezért a hidrogén-jodid képzödésének sebessége:
v1 = ![]() ,
,
ahol a szögletes zárójelben a koncentrációk szerepelnek; a k1-sebességi állandó pedig az egységnyi koncentráció mellett mért reakciósebességgel azonos. A hidrogén-jodid bomlására a visszahaladó reakcióban is felírhatjuk a reakciósebességet:
2 HI(g) = H2(g) + I2(g)
v2 = ![]() ,
,
mely reakció lejátszódásához ugyancsak két molekula ütközése szükséges. Ezért a sebességi egyenletben a hidrogén-jodid koncentrációjának a négyzete szerepel. A hidrogén-jodid képzödése és a bomlása is kinetikusan másodrendü reakció, mivel a sebesség a két reagáló anyag koncentrációjának a szorzatával (itt négyzetével) arányos. Mindkét folyamat bimolekulás, ennek ellenére hangsúlyozni kell, hogy a reakció rendüsége és molekularitása eltérhet egymástól. A koncentrációk hatványkitevöinek összege definíció szerint megadja a reakciók kinetikus bruttó rendjét.
v = k [A]a [B]b [C]c, a reakció kinetikus bruttó rendje a+b+c,
mely az A anyagra a-ad rendü, a B anyagra b-ed rendü, a C anyagra c-ed rendü. A reakció részrendjei elméleti megfontolások alapján nem számolhatók ki, azokat csak kísérletileg lehet meghatározni.
A kémiai reakciók részrendjei (a,b,c, ...) és bruttó rendüsége nem feltétlenül egész számokkal fejezhetök ki. Heterogén fázisú reakciók esetén egyébként még negatív részrendek is elöfordulnak. A pozitív egész számoktól eltérö rendüség általában bonyolult reakciómechanizmusra utal.
Reakciókinetikai szempontból különlegesek azok a reakciók, melyeknek sebessége nem koncentrációfüggö (nulladrendü reakciók), tehát sebességi egyenletük:
v = k.
Elsörendü reakciók
A kinetikusan elsörendü reakciók sebessége egyetlen anyag koncentrációjától függ:
v = k [A].
Mivel a koncentráció mértékegysége
mol/dm3, a reakciósebességé pedig mol dm-3 s-1, ezért a k egysége s-
Ennek meg
![]() k [A].
k [A].
Ha a fenti differenciálegyenletben szereplö változókat szétválasztjuk, majd integrálást végzünk, akkor:
ln [A]o = ln [A] + kt,
ahol az átalakuló anyag kezdeti koncentrációja t = 0 idöpontban [A]o és t-idöpontban [A]. Az egyenlet átalakításával:
ln ![]() kt
kt
k = ![]() ;
;
tízes alapú logaritmusban kifejezve pedig:
k =
![]() .
.
A sebességi állandó tehát számítható, ha ismerjük a kezdeti koncentrációt, és ha mérjük a t idöpontig átalakult anyag mennyiségét. Elsörendü reakciókban az átalakuló anyag koncentrációja exponenciálisan csökken az idö függvényében. Természetesen a log[A] - idö függvénye viszont lineáris minden elsörendü folyamatra. Ilyen elsörendü folyamat pl. a szén-dioxid bomlása magas hömérsékleten szén-monoxiddá és oxigénné, valamint a ciklobután átalakulása eténné.
CO2(g) = CO(g) + O(g) k = 1,8 · 10-13s-1,
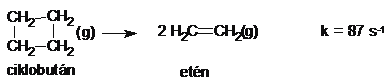
Ugyancsak elsörendü folyamat a nitrogén(V)-oxid (N2O5) bomlása gázfázisban, mely nitrogén-dioxidot és oxigént eredményez.
2 N2O2(g) = 4 NO2(g) + O2(g).
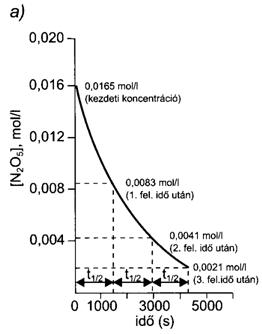
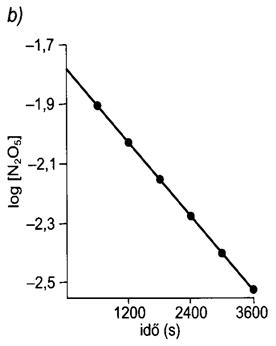
55. ábra. Az N2O5 bomlása gázfázisban 45 oC-on.
[A reakció felezési ideje független a kezdeti koncentrációtól; minden felezési idö eltelte után a koncentráció a felére csökken (a).A koncentráció logaritmusának idöbeli változása egyenest ad. (b)]
A felezési idö az az
idötartam, amely alatt az átalakult anyag koncentrációja a kezdeti érték
felére csökken. Ebben az idöpontban (t1/2) [A]=![]() , így a felezési idö kinetikusan
elsörendü reakciókban:
, így a felezési idö kinetikusan
elsörendü reakciókban:
t1/2
= ![]() ,
,
t1/2 = ![]() .
.
A kinetikusan elsörendü reakciókban a felezési idö független a kezdeti koncentrációtól, ezért a kémiai folyamat a sebességi állandó mellett a felezési idövel is pontosan jellemezhetö.
Másodrendü reakciók
A kinetikusan másodrendü reakciók sebessége a koncentráció második hatványával, vagy két koncentráció szorzatával arányos:
v = k [A]2, v = k [A]· [B].
A hidrogén-jodid bomlása gázfázisban másodrendü reakció:
2 HI(g) = H2(g) + I2(g) k = 2,4·10-21 dm3·mol-1·s-1.
A sebességi állandót a következöképpen származtathatjuk:
k = ![]() .
.
Az elsörendü reakcióknál leírt módon elvégezhetjük a másodrendü reakciók sebességi egyenletének integrálását. Az integrált sebességi egyenlet, ha a két anyag koncentrációja azonos, az alábbi:
v = k [A]2 vagy v = k [A]· [B] [A] = [B]
![]() kt.
kt.
Ez esetben a felezési idö fordítottan arányos a kezdeti koncentrációval:
t1/2
= ![]() .
.
Amennyiben másodrendü reakcióknál a koncentrációk különböznek ([A] [B]), akkor a sebességi egyenlet:
v = k [A]·[B] [A] [B]
ln ![]() kt,
kt,
és a felezési idö itt is fordítottan arányos a kezdeti koncentrációval.
A bimolekulás reakciók többnyire kinetikusan másodrendüek, azonban ha az egyik anyag koncentrációja igen nagy, vagy gyakorlatilag nem változik a másikhoz képest, akkor a reakció kinetikusan elsörendüvé válik. Ilyen pl. a szacharóz hidrolízise, ahol a hidrolízis sebessége csak a szacharóz koncentrációjától függ:
C12H22O11(aq) + H2O(l) = C6H12O6(aq) + C6H12O6(aq)
szacharóz glükóz fruktóz
v = k' [H2O] [C12H22O11] = k [C12H22O11].
A víz gyakorlatilag állandó koncentrációja a sebességi állandóval összevonható:
k = k' [H2O].
Nulladrendü reakciók
Nulladrendü reakciók sebessége független a koncentrációtól:
v = k [A]o = k (mivel [A]o = 1 ),
ezért az ilyen folyamatok állandó sebességgel játszódnak le; a folyamat sebességét nem az ütközések, hanem pl. a katalizátor felszínének aktív helyei befolyásolják. Ilyen folyamat pl. az ammóniagáz bomlása volfrámkatalizátor felszínén: W katalizátor
2 NH3(g) = N2(g) + 3 H2(g).
1000 oC
Nulladrendü folyamat integrált
sebességi egyenlete: [A] = [A]o - kt, felezési ideje pedig: ![]() , melyet csak a kiindulási anyag
mennyisége határoz meg.
, melyet csak a kiindulási anyag
mennyisége határoz meg.
A hömérséklet és a reakciósebesség
A reakciók sebessége növekszik a hömérséklet emelkedésével. Arrhenius szerint a reagáló részecskék megfelelö energiával, aktiválási energiával kell rendelkezzenek ahhoz, hogy a kémiai reakció lejátszódjon. Az Arrhenius-egyenlet az alábbi összefüggést adja a hömérséklet (T), az aktiválási energia (Ea) és a reakciósebességi állandó (k) között:
k = A e-Ea / RT ,
ahol A a reakcióra jellemzö ún. Arrhenius konstans,
R az egyetemes gázállandó.
A sebességi állandó logaritmikusan növekszik a hömérséklet emelkedésével. Fenti egyenlet mindkét oldalának tízes alapú logaritmusát véve az alábbi összefüggéshez jutunk:
log
k = log A - ![]() .
.
A sebességi állandó átlagosan 10 oC hömérséklet-emelkedés hatására 2-4-szeresre nö. A hömérsékletnek a reakciósebességre gyakorolt hatását az ütközési elmélet értelmezi.
Ütközési elmélet
A kémiai átalakulásokhoz a reagáló anyagoknak ütközniük kell egymással. Minden egyes ütközés magában hordozza a kémiai átalakulás lehetöségét, de nem minden ütközést követ kémiai reakció. A hidrogén-jodid gázmolekulák bomlási reakciója nagyon lassú, annak ellenére, hogy a másodpercenkénti ütközések száma majdnem azonos az ózon és a nitrogén-monoxid gázelegyében tapasztaltakkal, mely viszont igen gyorsan játszódik le:
O3(g) + NO(g) = O2(g) + NO2(g) k = 2,2·107 dm3 · mol-1 · s-1.
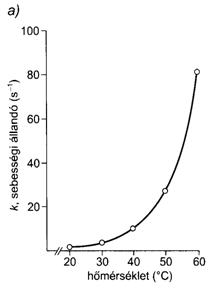
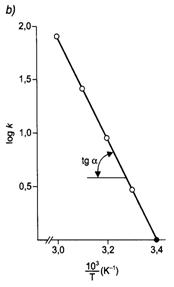
56. ábra. Kémiai reakciók sebességének változása a hömérséklettel.
[A sebességi állandó változása a hömérséklettel (a), és a sebességi állandó logaritmusának változása az abszolút hömérséklet reciprokának függvényében (b)]
Az ütközés szükséges feltétele ugyan a kémiai reakciónak, de nem minden ütközés vezet átalakuláshoz. Csak azok az ütközések hatásosak, amelyeknél az átlagosnál nagyobb energiájú részecskék találkoznak, vagyis azok amelyeknél rendelkezésre áll az aktiválási energia. A reakciók többségénél az aktiválási energia a hömozgásból származik. Az aktiválási energia azt az energiatöbbletet jelenti, amellyel a molekulák egy része rendelkezik az átlaghoz képest, ami elegendö ahhoz, hogy a molekula átalakuljon. A Maxwell-Boltzmann sebességeloszlási görbékkel értelmezhetjük az aktiválási energia szerepét a kémiai reakciókban. A hömérséklet emelkedésével nö azon molekulák száma, amelyek az aktiválási energiánál nagyobb energiával rendelkeznek. Nö a reakciósebesség, mivel az ütközésekben nagyobb valószínüséggel találkoznak aktivált molekulák. Magas hömérsékleten a molekulák nagyobbik hányada rendelkezik aktiválási energiával, ezért a reakció igen gyorssá válik. A kémiai folyamatok aktiválási energiaigénye igen eltérö. 75-150 kJ/mol az aktiválási energiája a szobahömérsékleten jól mérhetö sebességgel végbemenö reakcióknak, tehát a molekuláknak csak igen kis része tekinthetö aktivált állapotúnak. Az ennél kisebb aktiválási energiájú folyamatok gyakorlatilag pillanatszerüen mennek végbe, mert adott hömérsékleten a molekulák jóval nagyobb hányada kerülhet aktív állapotba, míg a nagyobb aktiválási energiát igénylö reakciók szinte mérhetetlenül lassúak is lehetnek, mert adott hömérsékleten a molekulák nagyon kis hányada éri el ezt a nagyobb aktiválási energiát.
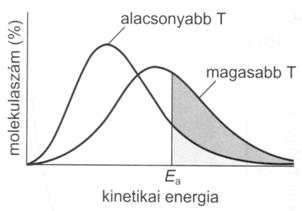
57. ábra. Hömérséklet hatása gázmolekulák kinetikus energiaeloszlására.
A vonalkázott terület jelenti az aktiválási energiánál nagyobb energiával rendelkezö részecskék számát)
Az Arrhenius-féle egyenlet linearizált formájának iránytangenséböl az aktiválási energia kiszámítható:
tg
a = - ![]() .
.
Hogy az ütközés kémiai átalakulással járjon, ahhoz az aktiválási energián kívül a molekulák megfelelö térbeli orientációja is szükséges. Ez különösen nagyméretü részecskék esetében jelentös. A sósav és az ammóniagáz reakcióját kismértékben befolyásolja a térbeli orientáció vagy sztérikus faktor. Az aktivált molekulák ütközése ugyanis csak akkor eredményes, ha az ammóniamolekula nitrogénatomja a sósavmolekula hidrogénatomja felöli részével ütközik. Ha az ammónia hidrogénjeit egy vagy több alkilcsoporttal helyettesítjük, akkor a sztérikus faktor szerepe még jelentösebbé válik. A tri-n-propil-amin sósavval történö reakciója esetében a nagyméretü szubsztituensek viszonylag kis helyet hagynak a sósav számára, ezért jelentösen csökken az eredményes ütközések lehetösége. Az ütközési elmélet értelmezi az Arrhenius-féle egyenletekben szereplö tényezöket. Az Arrhenius-konstans a másodpercenkénti ütközések száma és a sztérikus faktor szorzatával adható meg, az aktiválási energia és a reakciósebesség hömérsékletfüggése pedig az Arrhenius-féle egyenlet hatványkitevöjében szerepel.
Az átmeneti állapot elmélete
Az ütközési elmélet nem magyarázza az aktiválási energia szerepét. Az átmeneti állapot elmélete szükségessé teszi az ütközö aktivált molekulák megfelelö térbeli orientációját, melynek következtében a reagáló részecskékböl termékek jönnek létre. Ezen elmélet szerint olyan aktivált komplex képzödik, amely energiában gazdag, és amelyben bizonyos kötések lazítottak. A kémiai kötések gyengülése a rezgési energiává átalakult aktiválási energia révén valósul meg. Az aktivált komplexben a kötések átrendezödnek, az energiában gazdag állapot új kötések kialakítását teszi lehetövé, melynek következtében energia szabadul fel, és termékek képzödnek. A nagyon rövid élettartamú aktivált komplex kimutatása, szerkezetének meghatározása csak igen kevés reakció esetében lehetséges. Ennek ellenére feltételezhetjük annak általános elöfordulását. Az ózon és a nitrogén-monoxid ütközése során pl. az aktiválási energia rezgési energiává alakulva gyengíti az oxigének közötti kötést az ózonban, elökészítve az egyik oxigénatom leszakadását. Ugyanakkor az elektronsürüség a nitrogén felé tolódik el, melynek során részleges kötés alakul ki az ózon egyik oxigénatomja és a nitrogén között. Közben lazul a nitrogén-monoxidban lévö N O kötés is; az aktivált komplex felbomlik és létrejönnek a termékek.
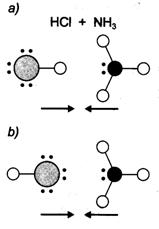
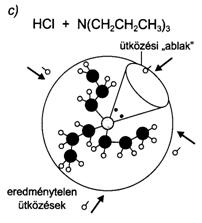
58. ábra. A sztérikus faktor szerepe a sósav reakciójában ammóniával, illetve tri-n-propil-aminnal.[Eredményes ütközés (a), eredménytelen ütközés (b) és az ütközési ablak (c) szerepe]

59. ábra. Az ózon és a nitrogén-monoxid reakciójában keletkezö
aktivált komplex szerkezete
Kémiai reakciók energiaváltozása
A termodinamikában megismertek alapján csak a szabadentalpia csökkenésével (DG < 0) járó reakciók mennek végbe spontán. Az aktiválási elmélet értelmében viszont csak a kellö mennyiségü aktiválási energiával rendelkezö molekulák alakulnak át. Ehhez a reakcióképes állapothoz, az aktivált komplex kialakításához, elöször energiát kell befektetni (Ea), majd a reakció lejátszódásakor az Ea felszabadul, és a szabadentalpia is csökken. A reakciósebesség és a termodinamikai törvények összekapcsolásával válik egyértelmüvé a termodinamikai spontaneitás és a reakciókinetikai végbemenetel közötti összefüggés. Az exergonikus folyamatok spontán játszódnak le, de sebességük akkor válik mérhetövé, ha az aktiválási energia az adott hömérsékleten kicsi. Az aktiválási energia nagysága tehát a reakció végbemenetelének gátja annak ellenére, hogy a befektetett Ea újra felszabadul és a reaktánsok számára újra felhasználhatóvá válik. A túl nagy aktiválási energia a termodinamikailag spontán lejátszódó folyamatokat is lehetetlené teszi. A szén-monoxid oxidációja pl. exergonikus folyamat, de az aktiválási energiája szobahömérsékleten olyan nagy, hogy az oxidáció nem megy végbe. Magasabb hömérsékleten elegendö energia áll rendelkezésre, ezért a termodinamikailag spontán folyamat reakciókinetikailag kellö sebességgel játszódik le:
CO(g) + O2(g) = CO2(g) + O(g) DGo = -25,5 kJ Ea= 213 kJ.
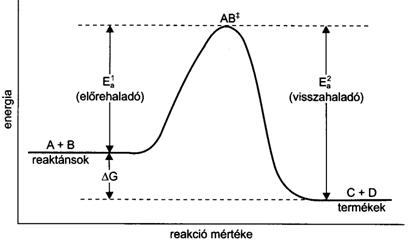
60.ábra.
Aktiválási energia szerepe a kémiai reakciók lejátszódásában. (E![]() az
elörehaladó, E
az
elörehaladó, E![]() a visszahaladó
folyamat aktiválási energiája; AB++ az aktivált komplex, DG a szabadentalpia-változás)
a visszahaladó
folyamat aktiválási energiája; AB++ az aktivált komplex, DG a szabadentalpia-változás)
Több elemi lépésböl álló reakciók
A bruttó folyamatokat leíró sztöchiometriai egyenletek nem adnak felvilágosítást az egymás melletti és utáni reakciólépésekröl. Az összetett reakciók több elemi lépésböl állnak, amelyek önállóan, de nem egymástól függetlenül játszódnak le. Az összetett reakciók legegyszerübb képviselöi az egyensúlyra vezetö folyamatok. Gyakoriak a konszekutív vagy sorozatreakciók, valamint az egyidejüleg lejátszódó párhuzamos reakciók.
Egyensúlyra vezetö reakciók
Az egyensúlyra vezetö reakciók során a termékek visszaalakulhatnak kiindulási anyagokká; a folyamat tényleges sebessége a két ellentétes irányú folyamat sebességének a különbsége. Egyensúlyra vezetö folyamat pl. a hidrogén-jodid képzödése:
![]() H2(g)
+ I2(g) 2 HI(g),
H2(g)
+ I2(g) 2 HI(g),
melynek során mindkét reakció kinetikusan másodrendü:
v1 = k1 [H2] [I2] és v2 = k2 [HI]2.
A folyamat tényleges sebessége:
v = v1 - v2 = k1 [H2] [I2] - k2 [HI] 2.
A v2 kezdetben 0, folyamatosan nö, a v1 pedig csökken. Egyensúlyi helyzetben v1 = v2 ekkor a bruttó folyamat (v = 0) gyakorlatilag megáll. Elvileg minden reakció megfordítható, reverzíbilis. A folyamat egyirányúsítását segíti elö a termék eltávolítása, vagy a nagy entalpiaváltozás.
A nitrogén-dioxid és a fluorgáz reakciója csak látszólag játszódik le egyetlen lépésben; bár a bruttó reakcióegyenlet alapján úgy tünik, mintha a reakció lejátszódásának elöfeltétele trimolekuláris ütközés lenne, azaz 2 NO2 és F2 molekula ütközne, aminek nagyon csekély a valószínüsége:
2 NO2(g) + F2(g) = 2 NO2F(g).
A felderített reakciómechanizmus alapján a folyamat tulajdonképpen két elemi reakcióban, bimolekulás ütközésekkel játszódik le:
NO2(g) + F2(g) = NO2F(g) + F(g) lassú lépés,
F(g) + NO2(g) = NO2F(g) gyors lépés.
Az elemi reakciók közül a második sokkal gyorsabb (k2 >> k1), ezért a folyamat bruttó sebességét a lassú, az elsö elemi reakció sebessége határozza meg. Általánosságban mindig a legkisebb sebességi állandójú elemi reakció határozza meg az egész folyamat sebességét.
Konszekutív vagy sorozatreakció
A sorozatreakciókban a termékek újabb reakcióba lépnek önmagukkal vagy más anyagokkal, és új termékek keletkeznek. Konszekutív pl. az alábbi általános átalakulás:
A
![]() B
B ![]() C
C
Az egyes lépések ebben az esetben kinetikusan elsörendüek, ezért sebességi egyenletekkel pontosan megadható a kiindulási anyag (A), a köztitermék (B) és a végtermék (C) koncentrációja, tetszés szerinti idöpillanatban. A kiindulási anyag (A) koncentrációja exponenciálisan csökken, a közti termék (B) koncentrációja gyorsan nö, majd egy maximum elérése után lassan csökken. Megfelelö k1/k2 arány mellett B koncentrációja viszonylag hosszabb idön át állandó maradhat. Ezt a viszonylag állandó köztitermék-koncentrációt egy egyensúlyi állapot hozza létre; idöegység alatt ugyanannyi B molekula keletkezik, mint amennyi elbomlik. Ez az állapot mindaddig fennáll, amíg a kiindulási anyag (A) koncentrációja kellöen nagy. Ez a közel egyensúlyi állapot alapvetöen különbözik a valódi egyensúlytól, amit az oda- és visszaalakuló reakciók alakítanak ki, ezért megkülönböztetésül a köztitermékek közel egyensúlyi állapotát stacionárius vagy steady-state állapotnak, míg viszonylag állandó koncentrációját stacionárius vagy steady-state koncentrációnak hívjuk. A végtermék (C) koncentrációja kezdetben igen lassan nö, mivel az A B átalakulás a sebesség meghatározó lépés, majd a C képzödési sebessége növekszik. Ilyen sorozatreakciónak tekinthetö a legtöbb biokémiai átalakulás az élö szervezetben.
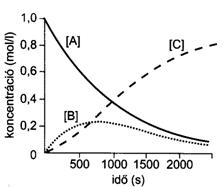
61. ábra. A koncentrációk változása sorozat reakciókban
Sorozatreakció például a biokémiában a glikolízis, melynek során a glikogénböl glükóz(1)-foszfáton keresztül glükóz(6)-foszfát, fruktóz(1,6)-difoszfát majd glicerin, a másik ágon pedig piroszölösavon keresztül tejsav ill. etil-alkohol keletkezik.
Konszekutív reakcióknak tekinthetö a citrátkör vagy más néven a trikarbonsav-ciklus is, melynek során az acetil csoport szene szén-dioxiddá oxidálódik, a hidrogének pedig hidrogénszállító koenzimekre tevödnek át. Konszekutív reakció ezen kívül pl. a zsírsavak b-oxidációja, melynek során a hosszú szénláncokból acetil-csoportok hasadnak le, amelyek aztán az acetil-CoA segítségével belépnek a citrátkörbe.
A fentiekben említett sorozatreakciók mechanizmusát használja fel Michaelis-Menten-elmélete az enzimkinetikában. Alapvetö különbség azonban az, hogy a Michaelis-Menten-elmélete az elsö lépést reverzíbilisnek tekinti:
![]() E
+ S ES E + P,
E
+ S ES E + P,
mely reakcióban E az enzim, S a szubsztrát és P a termék. Az ES köztitermék aktivált enzim-szubsztrát komplexnek felel meg, melynek koncentrációja a steady-state állapotnak köszönhetöen állandó.
Párhuzamos reakciók
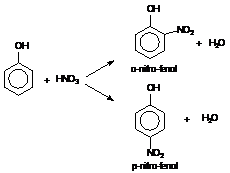 Ugyanaz az
anyagféleség ugyanabban a reakcióközegben különbözö kémiai reakcióban
vehet részt. Ezekben az esetekben párhuzamos vagy szimultán reakcióról
beszélünk. A párhuzamos reakciók nagyon gyakoriak a szerves kémiában.
Jellemzö példa erre a fenol nitrálása, amikor párhuzamos reakcióban orto-
és para-nitrofenol képzödik.
Ugyanaz az
anyagféleség ugyanabban a reakcióközegben különbözö kémiai reakcióban
vehet részt. Ezekben az esetekben párhuzamos vagy szimultán reakcióról
beszélünk. A párhuzamos reakciók nagyon gyakoriak a szerves kémiában.
Jellemzö példa erre a fenol nitrálása, amikor párhuzamos reakcióban orto-
és para-nitrofenol képzödik.
62. ábra. Az orto- és para-nitro-fenol képzödése
A párhuzamos reakciók sebessége igen eltérö lehet egymástól. A nagy sebességgel lejátszódó folyamatot föreakciónak, a többit pedig mellékreakciónak nevezzük. A reakciókörülmények megváltoztatásával a párhuzamos reakciók sebességi viszonyai megváltoztathatók, azaz a föreakció mellékreakcióvá, a mellékreakció föreakcióvá léphet elö. A biológiai rendszerekben is számos párhuzamos reakció játszódik le. A glikolízis példájánál maradva a fruktóz(1,6)-difoszfátból dihidroxi-acetonfoszfát vagy gliceraldehid-3-foszfát is keletkezhet. Az utolsó lépések egyikében a piroszölösavból keletkezhet tejsav, vagy keletkezhet belöle pl. acetaldehid is.
Az oxálacetát a biokémiai folyamatok során beléphet a citrátciklusba, a glükoneogenezis folyamatába és egyéb szintézisekben is részt vehet. A többféle átalakulásra képes metabolitok párhuzamos reakcióinak módosítása sokféle mechanizmussal történhet attól függöen, hogy milyen a szimultán reakciókat katalizáló enzimek aktivitása. A különbözö reakciók egymástól sokszor igen eltérö sebességgel mennek végbe. Hasonló párhuzamos reakciók mennek végbe a több szubsztráttal rendelkezö enzimek szimultán reakciói során is. Ezeket a reakciókat a biokémia részletesen tárgyalja.
Láncreakciók
Sok olyan kémiai folyamat van, amelyek sebessége lényegesen nagyobb, mint amit az aktiválási energia alapján várnánk. Az égések és robbanások csak bonyolult sebességi egyenletekkel írhatók le, melyek kinetikus rendje rendszerint nem egész szám. Reakciómechanizmusuk bonyolult, sok elemi lépésböl állnak; ezek az úgynevezett láncreakciók. A láncreakciók kialakulásának elsödleges feltétele, hogy a kezdeti reakció során olyan termékek jöjjenek létre, melyek igen kicsiny aktiválási energiával további reakciókra képesek. Ezen további reakciók során a termék újraképzödik, a kis aktiválási energiájú lépések fennmaradnak. A láncreakció addig folytatódik, amíg a láncreakcióra képes termékek újratermelödnek; amikor ezek eltünnek a rendszerböl, a láncreakció leáll. A durranógáz robbanásának reakcióját az alábbiak szemléltetik:
H2 = H. + H . lánckezdet,
H. + O2 + H2 = H2O + HO. láncfolytatás,
HO. + H2 = H2O + H. láncfolytatás,
H. + O2 = HO . + O. láncelágazás,
O. + H2 = HO. + H. láncelágazás.
A láncreakciót a hidrogénatomok képzödése vezeti be, a láncfolytatásban a gyökök (H· és HO·) újratermelödnek. A reakcióban láncelágazás is bekövetkezik, kettö-kettö gyök kialakulásával. A láncreakció befejezödése, letörése során gyökök nem képzödnek.
HO . + H . = H2O láncvégzödés,
H. + H . = H2 láncvégzödés.
Katalízis
A katalizátorok a kémiai reakciók sebességét meggyorsítják. A katalizátorok megváltoztatják a reakció mechanizmusát, új reakciókat nyitnak meg, miközben csak átmenetileg vesznek részt a reakcióban, valamely kiindulási anyaggal kisebb aktiválási energiájú átmeneti komplexet alkotva. A katalizátor csak a termodinamikailag végbemehetö folyamatok sebességét gyorsítják, nem befolyásolják azonban az egyensúlyi állapotot; az elörehaladó és visszahaladó reakciók sebességét azonos módon fokozzák. A katalizátor jelenléte tehát csökkentve az aktiválási energiát új reakcióutat nyit meg, mindkét irányba fokozza a kémiai reakciók sebességét, de nem befolyásolja a folyamat szabadentalpia változását és egyensúlyi állandóját.
Homogén katalízis
Homogén katalízisre példa a hidrogén-peroxid bomlása, melynek során a reagáló anyagok azonos fázisban vannak, és amely szobahömérsékleten lassú folyamat:
2 H2O2(aq) = 2 H2O(l) + O2(g) DGo = -94,7 kJ.
A bomlás sebességét jodidionok jelenléte fokozza. A katalízis eredményeként a folyamat aktiválási energiája jelentösen csökken. A katalizált reakció mechanizmusa az alábbi:
H2O2(aq) + I = H2O(l) + IO
IO + H2O2(aq) = H2O(l) + O2(g) + I
A katalízis során a jodidionok részt vesznek a folyamatban, a reakció második lépésében azonban újraképzödnek, így mennyiségük gyakorlatilag nem változik. A katalizáló hatás - egy meglehetösen szük tartományban - arányos a katalizátor koncentrációjával.
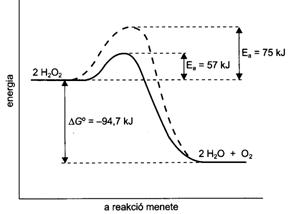
63. ábra. A hidrogén-peroxid bomlása nem katalizált (szaggatott görbe) és jodid ionokkal katalizált (folyamatos, vastag görbe) reakcióban
Heterogén katalízis
Heterogén katalízis során a katalizátor és a katalizált reakcióban résztvevö anyagok más fázisban vannak. Jelentösek a szilárd halmazállapotú katalizátorok, melyek müködése gyorsítja a gáz- és folyadékfázisban lévö anyag átalakulását. A katalizátor anyagi minösége, a különbözö kísérleti körülmények jelentösen befolyásolhatják a képzödö termékek minöségét. A katalizátorok szinte irányítják a kémiai folyamatokat, mert ugyanabból a kiindulási elegyböl más és más termékek képzödését segítik elö. A katalizátorok irányító hatását a szén-monoxid és a hidrogén reakciójában a 33. táblázatban lévö összeállítás mutatja.
33. táblázat. A katalizátor irányító hatása a szén-monoxid és a hidrogén reakciójában
|
Katalizátor |
Körülmények |
Termékek |
|
Ni |
100-200 oC, 0,1-1 MPa |
CH4 + H2O |
|
ZnO/Cr2O3 |
400 oC, 50 MPa |
CH3OH + H2O |
|
Co/ThO2 |
190 oC, 0,1-2 MPa |
CH4, C2H6, C3H8 + H2O |
|
Ru |
200 oC, 20 MPa |
Nagy molekulatömegü szénhidrogének + H2O |
|
ThO2 |
400 oC, 20 MPa |
Elágazó láncú szénhidrogének + H2O |
A 33. táblázatból látható, hogy a katalizátor és a reakciókörülmények megváltoztatásával a szén-monoxidból és a hidrogénböl egészen eltérö összetételü és molekulatömegü termékek képzödhetnek. A katalizátor felületén lévö aktív helyhez a reagáló anyagok adszorpcióval kötödnek. A kapcsolatot elsösorban a gyenge kölcsönhatások hozzák létre, amelyek végül is adszorpciót eredményeznek. A kémiai adszorpció vagy kemiszorpció a London-féle eröknél lényegesen erösebb kölcsönhatás, ezért itt a reaktánsokban lévö kémiai kötések fellazulnak. A heterogén katalízis lépései ill. azok sebessége igen eltérö lehet. A reagáló anyagok diffúziója a katalizátor felületére általában lassú folyamat, az adszorpció vagy kemiszorpció igen gyors, és ugyancsak gyors a tényleges kémiai reakció a katalizátor felületén. A termékek deszorpciója lehet gyors is, míg a termékek diffúziója lassú.
A hömérséklet és a nyomás növelése megnöveli a diffúziós folyamatok sebességét, ami a katalízis szempontjából kedvezö. Ugyanakkor nagymértékben befolyásolja az adszorpciót és a deszorpciós folyamatot.
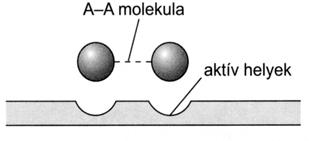
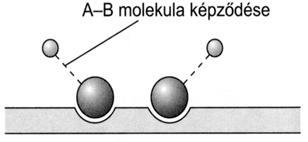
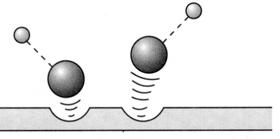
64. ábra. A heterogén katalízis mechanizmusa az A-A + 2 B = 2 A-B reakcióban. (Szilárd katalizátor aktív helyeihez kötödö A-A molekula kovalens kötései
fellazulnak, a katalizátor felületén A-B molekulák képzödnek, majd a
termék deszorpciójával a katalizátor regenerálódik)
E két paraméter kísérleti tapasztalatokkal alátámasztott optimális megválasztása alakítja ki a katalizált reakció optimális körülményeit. Idegen anyagok jelentösen módosíthatják a heterogén katalizátorok aktív helyeit. Rendkívül kis koncentrációban képesek a katalizátor hatékonyságát megszüntetni, vagy jelentösen csökkenteni. Az ilyen anyagokat katalizátormérgeknek vagy inhibitoroknak nevezzük. Hatásuk abból áll, hogy a katalizátorral úgy lépnek reakcióba, hogy a katalizátor müködése lecsökken vagy megszünik. Az aktivátorok fokozzák a katalizátor hatékonyságát oly módon, hogy a katalizátor felületén rácshibákat alakítanak ki, amelyek gyakran azonosak az aktív helyekkel.
Heterogén katalizátorok vannak a gépjármüvek kipufogórendszerében, melyek a környezetre rendkívül káros szén-monoxid és nitrogén-monoxid gázokat ártalmatlan szén-dioxiddá ill. nitrogéngázzá alakítják át. A különbözö üzemanyagok ólomtartalma rendkívül erös katalizátorméreg, ezért a katalizátoros gépkocsikhoz csak ólommentes benzin használható.
Az élö szervezet katalizátorai, az enzimek is sok esetben heterogén katalízist valósítanak meg. E biokatalizátorok esetén lényeges különbség az, hogy elsösorban csak egy reakciót képesek katalizálni; aktív helyük föleg a szubsztrát megkötését teszi lehetövé. A reakció aktiválási energiáját rendkívüli mértékben csökkentik. A hidrogén-peroxid spontán bomlásakor pl. az Ea= 75 kJ, jodidion katalízis esetén Ea= 57 kJ, heterogén katalízis során platina alkalmazásakor Ea= 49 kJ, kataláz enzim jelenlétében pedig Ea= 23 kJ. Az enzim müködését itt is módosíthatják aktivátorok ill. inhibitorok. Az enzimreakciók müködését, valamint az aktivátorok ill. inhibitorok hatását a biokémia megfelelö fejezetei tárgyalják.
Találat: 6531