 | |||||||||
|  | ||||||||
| |||||||||
| | ||||||||
| kategória | ||||||||||
|
|
||||||||||
|
|
||
HOMOGÉNKATALITIKUS AMINOKARBONILEZÉS: ÚJ MÓDSZER KIDOLGOZÁSA N-ferrocenoil-aminosav-észterek, FERROCÉNGLIOXAMIDOK ÉS DISZUBSZTITUÁLT FERROCÉNSZÁRMAZÉKOK SZINTÉZISÉRE
1.2. A ferrocénkarbonsavamidok tulajdonságai, felhasználásuk
1.2.1. Molekula- és ionreceptorok
1.3. Ferrocén-amidok elõállítása
1.4. N-Ferrocenoil-aminosavak és -peptidek elõállítása
1.5. Palládium-katalizált reakciók
1.5.1. A jód-ferrocén és a dijód-ferrocén palládium-katalizált reakciói
2. Az eredmények bemutatása és értékelése
2.2. Jód-ferrocén aminokarbonilezése aminosav-észterek jelenlétében
2.3. 1,1'-dijód-ferrocén aminokarbonilezése különbözõ szekunder aminok jelenlétében
2.5. 1,n'-ferrocéndikarboxamidok szerkezetének vizsgálata
2.6. 1,1'-dijód-ferrocén aminokarbonilezése aminosav-észterek jelenlétében
2.7. Heterodiszubsztituált 1,1'-ferrocén származékok elõállításának egy további lehetõsége
3.1. A kísérleti munka során felhasznált anyagok elõállítása, minõsége
3.1.1. Alapanyagok, katalizátorok és segédanyagok
3.2. A kísérletek kivitelezése, az egyes származékok kinyerése
3.2.1. Jód-ferrocén elõállítása
3.2.2. 1,1'-dijód-ferrocén elõállítása
3.2.3. Aminosav-metil-észterek elõállítása
3.2.4. Jód-ferrocén karbonilezése aminosav-észterek jelenlétében
3.2.5. 1,1'-dijód-ferrocén aminokarbonilezése aminok jelenlétében
3.2.6. 1'-jód-ferrocénkarboxamid és 1'-jód-ferrocénglioxamid Stille-reakciója
3.2.7. 1'-benzoil-N,N-dietil-ferrocénkarboxamid (15a) elõállítása
3.2.8. 1,1'-dijód-ferrocén aminokarbonilezése aminosavészterek jelenlétében
3.2.9. 1'-acetil-N-ferrocenoil-glicin-metil-észter (18) elõállítása
3.3. Mûszeres analitikai vizsgálati módszerek és készülékek
4. Az elõállított vegyületek analitikai adatai
A ferrocénszármazékok jó stabilitásuknak és kedvezõ elektrokémiai tulajdonságaiknak köszönhetõen gyakorlati szempontból nagy jelentõséggel bírnak. Felhasználhatóak homogénkatalitikus szintézisekben ligandumként, redox aktív molekula- és ionreceptorként, redoxkapcsolóként, vagy bioszenzorként. A ferrocénkarbonsav amid származékainak jelentõségét figyelembe véve fontos lehet egy új, egyszerû, ugyanakkor hatékony módszer kidolgozása ezen vegyületek szintézisére.
A munka célja N-ferrocenoil-aminosav-észterek, a ferrocénvázas vegyületek között újszerû ferrocénglioxamidok, valamint egyéb ferrocénszármazékok szintézise volt, a ferrocénszármazékok elõállítása során eddigiekben nem alkalmazott palládium katalizált homogénkatalitikus karbonilezési eljárással.
A Pannon Egyetem Szerves Kémia Intézeti Tanszékén elkezdett jód-ferrocén karbonilezési kísérletek során új típusú ferrocénglioxamidokat szintetizáltak, ferrocénkarboxamid melléktermék keletkezése mellett.
A munka folytatásaként a szerzõ vizsgálta a jód-ferrocén palládium katalizált aminokarbonilezését aminosav-észterek mint nukleofil reakciópartnerek jelenlétében.
Megállapította, hogy a korábbi jód-ferrocén és szekunder aminok karbonilezési reakcióinál tapasztaltakkal ellentétben nem a hõmérséklettel, hanem a megfelelõ bázis alkalmazásával lehet a reakciók szelektivitását eredményesen befolyásolni. A vizsgálatok eredményeibõl meghatározta, a ferrocénglioxamidok, illetve ferrocénkarboxamidok elõállításához optimális reakciókörülményeket. Bizonyította, hogy karbonilezési körülmények között a bázisként alkalmazott DBU is acilezhetõ.
Az 1,1'-dijód-ferrocén aminokarbonilezésének vizsgálata rámutatott, hogy a megfelelõ reakciókörülmények megválasztásával 1'-jód-ferrocénkarboxamidok és -glioxamidok közepes hozammal szintetizálhatóak, melyek további homogénkatalitikus reakciókba vihetõk, változatos szerkezetõ heterodiszubsztituált ferrocénszármazékokat eredményezve.
A
részletes kutatómunka eredményeként egyszerû és hatékony eljárás született két
különbözõ amidocsoportot tartalmazó 1,1'-ferrocéndikarboxamidok, illetve 1,n'-ferrocenoil-aminosav-észtereket
elõállítására, melyekre rendkívül kevés példát találunk az irodalomban.
A ferrocén felfedezésével, szerkezetének tisztázásával új korszak nyílt meg a fémorganikus kémia területén. Számos eljárást dolgoztak ki a legkülönbözõbb ferrocénszármazékok szintézisére, mely egy olyan dinamikusan fejlõdõ vegyületcsalád megismerését jelentette, amely gyakorlati szempontból rendkívül hasznos tulajdonságokkal rendelkezik. Használják õket homogénkatalitikus szintézisekben ligandumként, redox aktív molekula- és ionreceptorként, redoxkapcsolóként, vagy bioszenzorként.
Az említett vegyületek közül az N-ferrocenoil-aminosavszármazékok biokémiai és farmakológiai szempontból is igen nagy jelentõséggel bírnak. Jól alkalmazhatóak például élõ szervezetekben lejátszódó elektrontranszfer folyamatok vizsgálatára, vagy különbözõ molekulákra érzékeny bioszenzorként. Emellett segítségükkel bizonyos peptidek szerkezeti jellemzõirõl is információt nyerhetünk.
Napjainkban a szerves molekulák szén-monoxiddal történõ funkcionalizálása dinamikusan fejlõdõ eljárás, alkalmazása várhatóan továbbra is teret hódít a szintetikus kémia területén.
A karbonilezési folyamatok tárgykörén belül nagyszámú, igen részletes irodalom áll rendelkezésre az aminok jelenlétében lejátszódó aminokarbonilezés leírására vonatkozóan.
A kiterjedt kutatások ellenére ferrocénszármazékok ily módon történõ elõállítását egyedül kutatócsoportunk vizsgálta.
Az irodalmi adatok azt mutatják, hogy az aminokarbonilezés eredménye rendkívül érzékeny a reakciókörülmények változásaira, legyen az akár a kiindulási anyagkánt használt szerves molekula, az alkalmazott bázis vagy a nukleofil reagensként használt amin, de a reakció hõmérséklete, a szén-monoxid nyomás, oldószer, katalizátor mind jelentõsen befolyásolják a karbonilezés kimenetelét.
A ferrocénkarbonsav amidszármazékai gyakorlati jelentõségének okán fontos lehet egy, az eddigieknél hatékonyabb és ugyanakkor egyszerûbb eljárás kidolgozása. Erre kínál lehetõséget az elõbb említett, különbözõ nukleofil reagensek jelenlétében végrehajtott aminokarbonilezési eljárás.
Kísérleteim kiindulási vegyületeként jód-ferrocént, illetve 1,1'-dijód-ferrocént, nukleofil reakciópartnerként primer és szekunder aminokat, illetve aminosav-metil-észtereket használtam. A vizsgálataim fõként arra irányultak, hogy új, hatékony eljárást dolgozzak ki ferrocénkarboxamidok és ferrocénglioxamidok elõállítására. Az elõállított új vegyületek elkülönítésén és karakterizálásán túlmenõen egy esetben a vegyület mélyreható szerkezetvizsgálatát is elvégeztem. Továbbá egyéb homogénkatalitikus eljárások kombinációjával új ferrocénszármazékok szintézisének lehetõségét is vizsgáltam.
Ebben a fejezetben szeretném összefoglalni munkám elõzményeit, irodalmi hátterét. Igyekszem bemutatni a dolgozat alapját képezõ ferrocénszármazékok tulajdonságait, felhasználási lehetõségeit, lehetséges elõállítási módjait. Röviden áttekintem az általam használt jód-ferrocén és 1,1'-dijód-ferrocén palládium-katalizált homogénkatalitikus reakcióit. Részletesebben tárgyalom az aminokarbonilezési reakciók általános jellemzõit.
A ferrocén felfedezése, szerkezetének megismerése, karakterizálása az 1950-es évek elején a fémorganikus kémia robbanásszerû fejlõdéséhez vezetett és e vegyületet a mai napig töretlen tudományos érdeklõdés övezi.
Elõször Kaely és
Pauson számolt be egy új típusú vas-organikus vegyületrõl a "Nature" hasábjain
1951-ben [1] Ciklopentadienil-magnézium-bromidot reagáltattak
vas(III)-kloriddal, azzal a céllal, hogy fulvalént ![]() állítsanak
elõ (1.1 egyenlet). Ehelyett egy meglepõen stabil, narancssárga színû port
kaptak. A stabilitást a ciklopentadienil-anion aromás jellegének tudták be.
állítsanak
elõ (1.1 egyenlet). Ehelyett egy meglepõen stabil, narancssárga színû port
kaptak. A stabilitást a ciklopentadienil-anion aromás jellegének tudták be.
![]() (1.1)
(1.1)
Tõlük függetlenül Miller és munkatársai szintén "diciklopentadienil-vas"-at szintetizáltak (1.2 egyenlet).
![]() (1.2)
(1.2)
A bisz(η5-ciklopentadienil)vas valódi szerkezetét õk még nem ismerték fel. Robert Burns Woodward és Goeffrey Wilkinson következtetett elõször a szendvics szerkezetre [2]. Tõlük függetlenül Ernst Otto Fischer szintén erre a megállapításra jutott.[3]. Feltételezésük helyességét NMR és röntgen adatokkal igazolták.
A koordinatív kapcsolat a fématom üres d-orbitáljainak és a ciklopentadienil-gyûrûk delokalizált π-MO-jainak átfedése révén jön létre.
Kémiai tulajdonságait tekintve a ferrocén - aromás jellegénél fogva - számos aromás szénhidrogénekre jellemzõ reakcióba vihetõ. A ferrocén reakciókészségét alapvetõen meghatározza az a tény, hogy a ciklopentedienil-gyûrû parciális negatív töltést hordoz, ezért a benzolszármazékokhoz képest elektrofil reagansekkel szemben nagyobb reakciókészséget mutat.
Az elektrofil szubsztitúciós reakcióban az elektrofil reakciópartner nem lehet oxidálószer, mert a Fe(II) → Fe(III) oxidációban keletkezõ ferricíniumion ( [Fe(Cp)2] ) elektrofilekkel nem támadható.
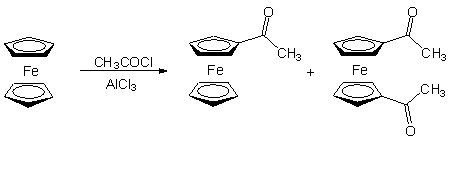
A Cp gyûrû nagy reakciókészségét mutatja, hogy Friedel-Crafts-acilezés során minden esetben termékelegyet kapunk. Például az alábbi reakcióban a monoacetil-ferrocén mellett megfigyelhetõ az 1,1'-diacetil-ferrocén keletkezése is [4].
A ferrocénkarbonsav amidszármazékai széles körben alkalmazott építõkövek a szupramolekuláris kémiában. Használják õket redox aktív molekula- és ionreceptorként, redox kapcsolóként, vagy bioszenzorként. Mindez a ferrocénvegyületek különleges szerkezeti és elektrokémiai tulajdonságainak köszönhetõ.
A ferrocénnek jól karakterizálható egy elektronos oxidációja, valamint mind a ferrocén, mind pedig a ferrocínium kation stabilitása teszi a ferrocén származékait különösen alkalmassá arra, hogy elektrokémiai szondaként számos területen alkalmazhatóak legyenek. A ferrocénszármazékok redox sajátságát a szubsztituensek erõsen befolyásolják. Ha ezekhez valamilyen vendég (guest) molekula, vagy ion kapcsolódik, az változást idéz elõ a ferrocén egység redox viselkedésében, ami például ciklikus-voltammetria segítségével nyomon követhetõ.
A dolgozat korlátait figyelembe véve csak néhány konkrét példát említek a számos felhasználási lehetõség közül.
A redox érzékeny receptorok olyan molekulák, amelyek képesek egy vendég (guest) molekula vagy ion szelektív megkötésére, amely azután nemkötõ kölcsönhatásokon, és/vagy különbözõ kötéseken keresztül hatást fejt ki a redox központra. A hatás elektrokémiai módszerekkel (általában ciklikus voltammetriával) mérhetõ.
Az amidszármazékok között találhatunk kationra, anionra, vagy neutrális molekulákra érzékeny receptorokat is.
Az elsõ csoportra kiváló példa a ferrocéndikarbonsav N,N-dialkil-amid származékai, melyek az alkáli-fémionok közül szelektíven csak a lítiumiont kötik meg az 1.4 egyenletben feltüntetett komplexek képzõdése révén. A komplexek kialakulását különbözõzõ analitikai módszerekkel (13C-NMR, CV, UV-Vis) igazolták [5].
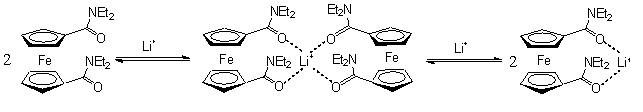
LiBF4 fokozatos adagolása az N,N-dietil-ferrocénkarboxamid oldatához a karbonil szénatom eltolódásának folyamatos növekedéséhez vezetett a 13C-NMR spektrumban, mivel a karbonil-oxigénhez koordinálódó Li+ növeli az oxigénatom elektronszívó hatását. A ciklikus-voltammogramon a LiBF4 adagolása a csúcspotenciál pozitív irányú eltolódását, illetve a csúcsáramok intenzitásának csökkenését, valamint nagyobb Li+ felesleg (>1ekv.) - mintegy 300mV-al nagyobb potenciálértéknél - egy újabb, a ferrocén-dikarboxamid/lítium=1/1 komplexhez tartozó potenciálcsúcs megjelenését eredményezte. Ugyanakkor Na+ és K+ ionok hozzáadása nem okozott detektálható változást a fenti vizsgálatok során.
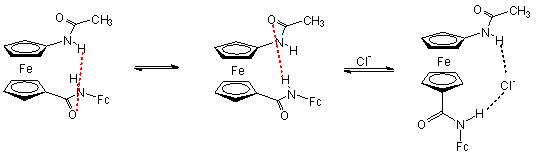
Az 1.5 egyenletben látható amidot kloridionokkal reagáltatták. Az anion hasítja a receptor molekula intramolekuláris hidrogénkötését, és egyidejûleg koordinálódik mindkét amidocsoporthoz hidrogénkötésen keresztül. A kloridion koordinációja megváltoztatja a két ciklopentadienil gyûrû elfordulásának szögét, valamint a ferrocén/ferrocínium redox pár redoxpotenciálját, így a kloridkoncentráció mérhetõ [6, 7].
Az Astruc és munkatársai által kifejlesztett, amidoferrocenil-alkiltiolát ligandummal módosított arany elektród (1.1 ábra) szelektíven ismeri fel a dihidrogén-foszfát aniont. A H2PO4- anion - hidrogénkötés-akceptor negatív töltésû oxigénje, illetve hidrogénkötés-donor savas OH csoportja miatt - két hidrogénkötésen keresztül is képes koordinálódni az ugyancsak kettõs tulajdonságú amidocsoporttal rendelkezõ receptor molekulához (1.1 ábra). Más szervetlen anionokkal végzett vizsgálatok (pl. HSO4-, NO3-, Cl-, és Br-) nem okoztak jelentõs változást a ciklikus voltammogramon [8].
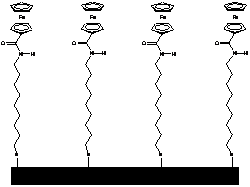
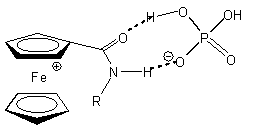
1.1 ábra Amidoferrocenil-alkiltioláttal módosított arany elektród és a H2PO4- anionnal kialakított hidrogénkötések
Ferrocéntartalmú amidopiridil receptorok 1,1'- és 1,3- regioizomerjei heterociklusos molekulákkal képeznek komplexeket komplementer hidrogénhidakon keresztül. A redoxaktív ferrocenilcsoport jelenlétének köszönhetõen a komplexképzõdés folyamata elektrokémiai módszerekkel szintén nyomon követhetõ [9, 10].
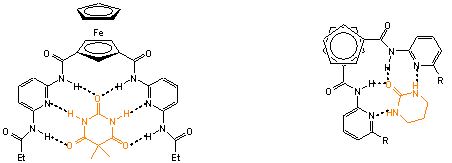
1.2 ábra Ferrocéntartalmú amidopiridil receptorok heterociklusos vegyületekkel alkotott komplexei
A bioszenzorok olyan mikró kivitelû elektrokémiai, optikai, vagy egyéb érzékelõk, melyek felületére biológiailag aktív anyagot kötnek. A detektált jel függvénykapcsolatban van egy mérendõ anyag mennyiségével.
A géndiagnosztika egyik kulcskérdése a DNS molekula érzékeny és egyszerû detektálásának lehetõsége. A legáltalánosabban alkalmazott vizsgálati módszer, hogy egy próba DNS-t, mely a célpont DNS komplementer szekvenciájával rendelkezik, radioaktív izotóppal, például 32P-al jelölnek meg és a két DNS molekula között kialakult kapcsolat miatt detektálhatóvá válik a keresett DNS szakasz. Habár a radioizotópos nyomjelzés a jó detektálhatóság miatt széles körben elterjedt, veszélyessége, illetve a jelzõ izotóp rövid élettartama miatt más alternatív megoldások válhatnak jelentõssé. Az egyik lehetõség az elektrokémiai módszerrel történõ detektálás. Habár a DNS közönséges körülmények között redox-inaktív, különbözõ módszerekkel, például ferrocenkarbonsav származékok hozzákapcsolásával aktívvá tehetõ (1.3 ábra). Az így kapott próba DNS által keresett DNS nemcsak hogy elektrokémiai úton jól detektálható, de mennyiségileg is meghatározható


1.3 ábra Ferrocén tartalmú próba DNS
Heller és Degani glükóz-oxidáz enzimek aminosav végeihez kapcsoltak ferrocén egységeket peptid kötéssel (1.4 ábra). Ezáltal közvetlen elektronikus kapcsolat jöhet létre egy elektród és az enzim FAD/FADH központja között, ami pusztán az enzim és az elektród között nem tud kialakulni. Azáltal, hogy a ferrocén/ferrocínium pár képes elektronokat közvetíteni az enzim és az elektród között, lehetõség nyílik az 1.4 ábrán látható enzimkatalizált reakcióban keletkezõ redukált állapotú FADH direkt reoxidálására. Ha az elektródra feszültséget kapcsolunk elektronvándorlás indul meg, a mérhetõ áram erõssége (I) függvénykapcsolatban van a glükóz koncentrációval [12].
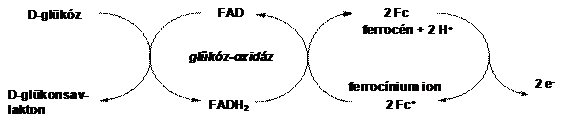
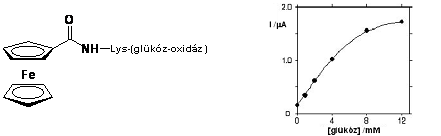
1.4 ábra Ferrocénnel módosított glükóz oxidáz enzim mûködése
A ferrocén felfedezését, karakterizálását követõen számos kutatócsoport irányította figyelmét ferrocénszármazékok, köztük a ferrocénkarboxamidok szintézisére. Az 1950-es évek második felére már több módszer is létezett az amidok elõállítására.
Weliky és Gould jó néhány, különbözõ ferrocénszármazékokhoz vezetõ reakció utat dolgozott ki benzoil-ferrocénen keresztül. E vegyület oximjának Beckman-átendezõdésével jutottak el a ferrocénkarboxanilidhez is (hozam 8%) [13].

Több N-szubsztituált ferrocénkarboxamidot állítottak elõ Rausch és munkatársai ferrocénbõl kiindulva. A ferrocént csekély feleslegben lévõ alumínium-kloriddal és a megfelelõ izocianáttal reagáltatva közepes hozammal nyerték a kívánt karboxamidokat [14].

Az izocianát ![]() amid átalakítás
másik érdekes esete, amikor nem ferrocénnel reagáltatják a megfelelõ
izocianátot, hanem lítium-ciklopentadieniddel. Ezután a
monokarbamoil-szubsztituált ciklopentadienid vas(II)-kloriddal lejátszódó reakciójából
kapják meg a monokarbamoil-, illetve dikarbamoil-ferrocént [15].
amid átalakítás
másik érdekes esete, amikor nem ferrocénnel reagáltatják a megfelelõ
izocianátot, hanem lítium-ciklopentadieniddel. Ezután a
monokarbamoil-szubsztituált ciklopentadienid vas(II)-kloriddal lejátszódó reakciójából
kapják meg a monokarbamoil-, illetve dikarbamoil-ferrocént [15].

(1.8)
Gyakran alkalmazott módszer a ferrocénkarbonsav kloridjával történõ acilezés. Némi hátrányt jelent azonban a savklorid nagy reakcióképessége ami megnehezíti a vegyület elõállítását, tisztítását, tárolását (ferrocénkarbonsavból számított hozam: 20-40% ) [16].
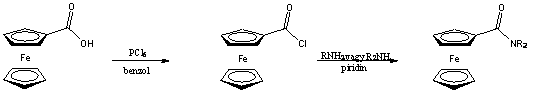
Ezt a hátrányt igyekezett kiküszöbölni Rotello azzal, hogy a szintézis intermediereként a megfelelõ savfluoridot használta, amely megfelelõen reaktív az amid képzéshez, ugyanakkor elég stabilis ahhoz, hogy tisztítása (átkristályosítás, szublimáció, kromatográfia) könnyen megoldható legyen, emellett szobahõmérsékleten bomlás nélkül eltartható [17].
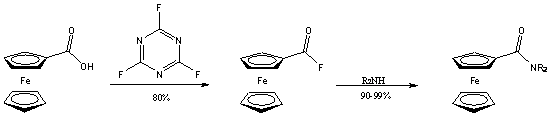
A savklorid és az amin kapcsolása általában bázis hozzáadását igényli, hogy az megvédje a támadó nukleofil reakciópartnert a protonálódástól. Meglepõ, hogy savfluorid esetében erre nincs szükség, ami további elõnyöket jelent használatakor. Elõször is, bázisra érzékeny szubsztrátum jelenlétében nem játszódik le mellékreakció, másodszor, nincs szükség vizes feldolgozásra a reakció végén.
Speiser azt vizsgálta, hogy különbözõ karbodiimidek hogyan képesek aktiválni a ferrocénkarbonsavat, hogy az erõs nukleofil reagenssel támadható legyen. A legtöbb vizsgált N,N'-diszubsztituált karbodiimid a ferrocénkarbonsavval igen stabil N,N'-diszubsztituált N-ferrocén-karbamid származékot eredményezett, kivéve az 1-etil-3-(3-dimetil-amino-propil)-karbodiimid-hidrokloridot (EDC). Éppen ezért alkalmas ez utóbbi karbodiimid a ferrocén-karbonsav aktiválására. Ferrocénkarbonsav, EDC és 3-(trietoxi-szilil)-propil-amin reakciójában közepes hozammal szintetizálták a megfelelõ ferrocénkarboxamidot
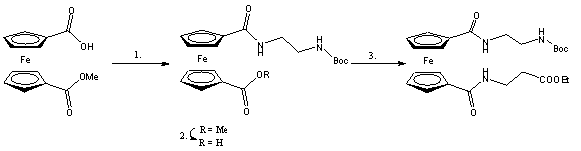
A leírt módszerek - 1,1'-ferrocén-dikarbonsavból kiindulva - természetesen alkalmasak 1,1'-ferrocéndikarboxamidok szintézisére is. Az 1,1'-heterodiszubsztituált ferrocéndikarboxamid származékok elõállítása azonban némileg bonyolult, hosszadalmas, többlépéses szintézist igényel azáltal, hogy a legtöbb esetben az egyik karboxilcsoportot védeni, míg a másikat aktiválni kell. A védõcsoport eltávolítását újabb aktiválási lépés követi és ezek után alakulhat ki a nem szimmerikus ferrocéndikarboxamid származék. Ez lehet az egyik oka, hogy ilyen vegyületek szintézisére csak elvétve találunk példát az irodalomban. Az egyik példa Sekine és munkatársai nevéhez fûzõdik. Ferrocéndikarbonsav-metil-észtert reagáltattak N-(t-butoxi-karbonil)-1,3-propándiaminnal 2-klór-1-metil-piridinium-jodid (CMPI) és tributil-amin jelenlétében (1.lépés) (hozam:89%). Majd a karbonsav-észter lúgos hidrolízise (2.lépés) után az elõzõvel megegyezõ módon alakították át a másik karboxilcsoportot 3-amino-propánsavval (1.11 egyenlet) (3.lépés) (hozam: 62%) [19].
Említést érdemelnek még ebben a fejezetben Kraatz és munkatársai, akik fõként N-ferrocenoil-aminosav-észterek, N-ferrocenoil-peptidek elõállításában és vizsgálatában alkottak maradandót, de ugyanazon módszerrel karboxamidokat is szintetizáltak (lásd következõ fejezet) [20].
A ferrocénkarbonsav amidszármazékai között különösen fontosak a N-ferrocenoil-aminosavak és -peptidek. Szolgálhatnak bioszenzorként (ld. 1.2.2. fejezet), de alkalmasak lehetnek a fehérjék bizonyos szerkezeti elemeinek modellezésére is.
Az elsõ ferrocéntartalmú aminosav- és dipeptid származékokat (Fc-CO-Gly-OMe, Fc-CO-Gly-OH, és Fc-CO-Gly-Leu-OEt) Schlögl állította elõ 1957-ben [21]. Ahhoz, hogy a peptidkötést enyhe körülmények között ki lehessen alakítani, a savat aktiválni kell. A ferrocenoil-aminosavak szintézise során a ferrocénkarbonsav aktiválására több megoldást is alkalmaztak (1.5 ábra):

1.5 ábra Ferrocénkarbonsav aktiválásának lehetõségei
Az aminosavak acilezésére alkalmas vegyület a karbonsav-klorid, melynek kialakítása oxaloil-kloriddal, vagy tionil-kloriddal történhet [21, 22].
További lehetõség a karbonsav átalakítása szukcinimiddé, vagy benzotriazol-észterré. Majd az aktivált ferrocénkarbonsav-származékot az aminosavészter oldatával 2 napig keverik (reakcióidõ: 2 nap, a nyerstermék hozama: 70-80%) [23].
A karbonsav
aktiválása történhet HBTU-val,  vagy TBTU-val
vagy TBTU-val is. Ez utóbbi megoldásnak több elõnye is van: a kívánt kapcsolás rövid idõ
alatt végbemegy, nem szükséges vízmentes oldószerek használata, megfelelõ
extraktív feldolgozással a kapcsoló reagensek eltávolíthatóak, ezáltal kiváló
hozammal, nagy tisztaságú terméket nyerhetünk (reakcióidõ: 30 perc, hozam:
52-87%) [24].
is. Ez utóbbi megoldásnak több elõnye is van: a kívánt kapcsolás rövid idõ
alatt végbemegy, nem szükséges vízmentes oldószerek használata, megfelelõ
extraktív feldolgozással a kapcsoló reagensek eltávolíthatóak, ezáltal kiváló
hozammal, nagy tisztaságú terméket nyerhetünk (reakcióidõ: 30 perc, hozam:
52-87%) [24].
Beck és munkatársai egy másik érdekes megoldást választottak az N-ferrocenoil-aminosav-észter elõállítására. A karbonsavat EDC-vel aktiválták és ezzel egy idõben hozzáadták az aminosav-észtert, emellett katalitikus mennyiségû 4-(dimetil-amino)-piridint (DMAP), valamint bázisként N-metil-morfolint használtak. A reakció hátránya a hosszú reakcióidõ (1-3 nap) [25] (hozam 73-78%).
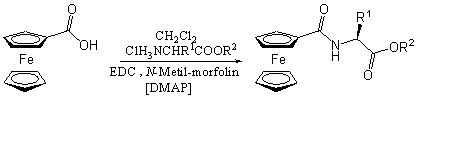
Az elõzõ fejezetben tárgyalt lítium-ciklopentadienid és izocianát reakcióján alapuló módszert alkalmazva szintén állítottak elõ ferrocenoil-aminosav-észtereket. Aminosav-észter-hidrokloridot feleslegben lévõ difoszgénnel reagáltattak, hogy megkapják a kívánt izocianátot, majd a karboxamidok elõállításánál leírt módon jutottak a mono-, illetve 1,1'-dikarbamoil-ferrocénhez [15].
Természetesen az itt tárgyalt módszerek mindegyike alkalmas 1,1'-diszubsztituált ferrocenoil-aminosav-észterek elõállítására is, a megfelelõ dikarbonsavból kiindulva.
Olyan 1,1'-heterodiszubsztituált ferrocenoil-aminosav származékok szintézisére, amikor két különbözõ aminosav, vagy peptid kapcsolódik a ferrocén két Cp gyûrûjéhez - a hasonló szerkezetû amidokhoz hasonlóan - jóval kevesebb példát találunk az irodalomban. Az általunk közzétett eredményeken kívül (melyeket a kísérleti részben ismertetek) mindössze három olyan publikáció született, amely a nem szimmetrikusan helyettesített 1,1'-ferrocén-dikarbonsav aminosav-, vagy peptid-származékait tárgyalja. Az egyiket Kimura és munkatársai tették közzé, akik sajnos a szintézis részleteit nem közölték [26]. A másik két közlemény Kraatz, illetve Metzler-Nolte nevéhez fûzõdik.
Kraatz az 1,1'-ferrocén-dikarbonsavat reagáltatta egyidejûleg Pro3-OBzl-rel és hidroxi-benzotriazollal (HOBt). Az így kapott termékelegybõl oszlopkromatográfiával elkülönítette a Fe[C5H4-CO-Pro3-OBzl][C5H4-CO-OBt]-t (hozam: 45- 55%). Majd következõ lépésként az OBt-csoportot cserélte a másik peptidre (Val-Val-Sta-Ala-Sta-OEt) (hozam: 55- 59%)[27].
Metzler-Nolte az 1,1'-ferrocén-dikarbonsavat HBTU és HOBt 1:1 arányú elegyével aktiválta, bázisként diizopropil-etil-amint használt. Ehhez adta a kétféle aminosavészter hidrokloridjának (Ala-OMe.HCl és Phe-OMe.HCl, illetve DPhe-OMe.HCl és Ala-OMe.HCl) a ferrocén-dikarbonsavra nézve fél ekvivalens, 1-1 arányú elegyét. Hat óra elteltével megismételte az aktivációs lépést, majd újabb adag aminosavészter hidroklorid elegy hozzáadását követõen egy teljes éjszakán át keverte (hozam: 29% és 23%). [28]
Az elmúlt évtizedekben az átmenetifém-organikus kémia fejlõdése a szerves szintézisutak forradalmi változását idézte elõ. Az ismert átmenetifém-organikus vegyületek közül a palládiumkomplexek váltak a legfontosabb katalizátorokká akár az alapvetõ nyersanyagok, a finomvegyszerek, vagy bonyolultabb, a természetben is elõforduló anyagok elõállítását vesszük figyelembe. A palládiumkatalizátorok jelenlétében lejátszódó átalakulások sokfélesége, a reakciók kitûnõ kemo-, regio- és sztereoszelektivitása lehetõvé teszi olyan rendkívül összetett molekulák szintézisének megvalósítását, amely a klasszikus szerves kémia módszereivel nem, vagy csak nagyon nehezen oldható meg.
A palládiumkatalizátorok többek között lehetõvé teszik, hogy egy molekula telítetlen szénatomján új szén-szén kötést hozzunk létre (Heck reakció-, vagy különbözõ fémorganikus reakciópartnerekkel lejátszódó kapcsolási reakciók segítségével), vagy C-heteroatom (C-N, C-P, C-S, stb) kötést alakítsunk ki a palládiumkomplex jelenlétében lejátszódó nukleofil szubsztitúció révén. További elõnyük, hogy képesek különbözõ kis molekulák, köztük a szén-monoxid aktiválására, mely így közvetlenül beépíthetõ valamely szerves molekulába. Az utóbbi folyamat karbonilvegyületek- vagy karbonsavszármazékok képzõdéséhez vezet.
A fenti reakciók kiindulási vegyületei a legtöbb esetben aril- vagy alkenil-halogenidek, illetve aril- vagy enol-triflátok, melyek a katalitikusan aktív palládiumkomplexszel reakcióba lépve szén-palládium kötést tartalmazó aril- vagy alkenil-palládium komplexek képzõdéséhez vezetnek. A központi palládiumatomhoz kapcsolódó telítetlen szénatomok a palládium "közvetítésével" reakcióba léphetnek különbözõ nukleofil reakciópartnerekkel (alkénekkel, fémorganikus reagensekkel, aminokkal, stb.). A halogénszármazékok közül - bár ezek a legdrágábbak - kitûnõ reakciókészségük miatt a jódvegyületeket alkalmazzák elsõsorban. Számos példa található azonban a brómszármazékok átalakítására is, az utóbbi idõben pedig egyre többen foglalkoznak a legkönnyebben hozzáférhetõ aril-kloridok reakcióival [29].
A bevezetésben említett reakciók esetében - legyenek azok egy elektrofil és nukleofil reakciópartner között végbemenõ kapcsolási reakciók, vagy szén-monoxid jelenlétében lejátszódó karbonilezési reakciók - az aktív katalizátor valamilyen Pd(0)-komplex. Az aktív részecske a reakcióelegyben alakul ki, a fõ kérdés általában az, milyen palládiumvegyületek hozzáadásával érhetjük el a legjobb eredményt. A leggyakrabban használt katalizátorrendszerek vagy már eleve 0-oxidációfokú palládium komplexet tartalmaznak, ilyen például a Pd(0)(PPh3)4 vagy a Pd(0)(dba)2 + nL rendszer, vagy olyan Pd(II)komplexek vagy Pd(II) sók, amelyekbõl a reakcióelegyben "in situ" alakul ki az aktív részecske valamilyen redukálószer (pl. egy amin, vagy a ligandumként használt foszfán) hatására. Az ilyen típusú katalizátorokra példa a PdX2L2 vagy Pd(OAc)2 + nL rendszer (L:egyfogú foszfán, X=Cl, Br, I).
A fejezet további részében a karbonilezési reakciókon kívül kizárólag azoknak a palládium-katalizált homogénkatalitikus reakcióknak a bemutatására szorítkozom, melyeket már alkalmaztak az általam is kiindulási anyagnak választott jód-, illetve 1,1'-dijód-ferrocén átalakítására. Jód-ferrocén és 1,1'-dijód ferrocén karbonilezését kutatócsoportunk valósította meg elsõ ízben. A munkám jelentõs részét képezõ karbonilezéssel kapcsolatos általános tudnivalókat az 1.5.2. fejezetben tárgyalom.
Bár a ferrocén reakcióiban az aromás vegyületekhez hasonló módon viselkedik, és az aril-halogenidek palládium-komplexek jelenlétében lejátszódó homogénkatalitikus reakcióit szívesen alkalmazzák aromás származékok szintézisére, a jód-ferrocén és dijód-ferrocén hasonló átalakítására viszonylag kevés példát találunk az irodalomban. A ferrocénszármazékok szintézisének legelterjedtebb kiindulási anyaga még mindig a ferrocénkarbonsav, illetve annak aktivált származékai.
Új szén-szén kötés kialakítása minden idõben a szintetikus szerves kémia meghatározó átalakításai közé tartozott. A klasszikus C-C kötést létrehozó módszerek (pl. Grignard reakció, Wittig reakció, cikloaddíció stb.) mellett az elmúlt 30 évben az átmenetifémek (Pd, Ni, Cu) által katalizált keresztkapcsolási (cross-coupling) reakciók jelentõsen kiterjesztették a szintetikus lehetõségeket.
A fémorganikus reagensek jelenlétében lejátszódó kapcsolási reakciók általában palládium(0) vagy nikkel(0) katalizálta reakciók. A kapcsolási reakció mechanizmusát leegyszerûsítve az 1.6 ábra mutatja be. A folyamat nyitó lépése egy szerves (általában halogén-) vegyület oxidatív addíciója az alacsony oxidációs állapotú átmenetifémre. A kapcsolni kívánt másik szerves molekularészlet a keletkezõ intermedierrõl ligandumcserével kerül az átmenetifémre. Általában ez a reakció a folyamat sebesség-meghatározó lépése. A transz elhelyezkedésû csoportok egy gyors izomerizációs lépés eredményeként cisz-helyzetbe kerülnek, majd a záró reduktív eliminációs lépés eredményeképpen kialakul a kapcsolt termék és a katalitikus ciklus bezárul az alacsony oxidációs állapotú átmenetifém újraképzõdésével.
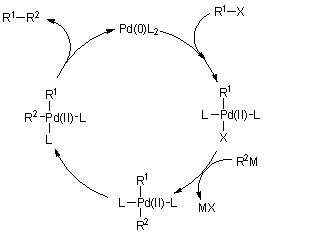
1.6 ábra A fémorganikus reakciópartnerek jelenlétében lejátszódó kapcsolás általános mechanizmusa
A leggyakrabban használt katalizátorok vagy már eleve 0-oxidációfokú palládiumot tartalmazó vegyületek, vagy olyan Pd(II)komplexek vagy Pd(II) sók, amelyekbõl a reakcióelegyben "in situ" alakul ki az aktív részecske valamilyen redukálószer (foszfán, fémorganikus reagens, trietil-amin) hatására.
Az elektrofil reakciópartner, amelynek oxidatív addíciójával a folyamat elindul, elsõsorban aril/alkenil-halogenid, vagy -triflát. A fémorganikus reakciópartnerek között találunk ón- (Stille kapcsolás) [30], bór- (Suzuki kapcsolás) [31-35], cink-, alumínium- és cirkónium- (Negishi kapcsolás) [36, 37], szilícium- és germánium- [38, 39] lítium- és magnézium- [40], valamint indiumvegyületeket [41].
A keresztkapcsolási eljárások legelterjedtebb és részleteiben is ismert képviselõje a Stille nevével fémjelzett reakció. A folyamatban a ligandumcsere az ónorganikus vegyület és a palládiumkomplex között játszódik le.
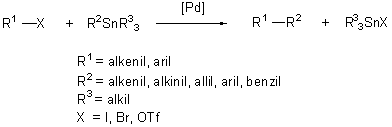
(1.13)
A kapcsolási reakcióban az ónatomon lévõ csoportok közül csak az egyik vesz részt. Szerencsére az ónról a legkülönbözõbb csoportok vihetõk át szelektíven, hiszen az alkil-transzfer sebessége jóval kisebb, mint a telítetlen csoportoké. Így az általánosan alkalmazott ónreagensek nem szimmetrikusak, három alkilcsoportot (metil-, vagy butil-) negyedikként pedig alkinil-, alkenil-, aril-, benzil- vagy allilcsoportot tartalmaznak. A módszernek számos elõnye van: a kereskedelmi forgalomban sokféle trialkil-ón származék megtalálható. E vegyületek általában kevéssé érzékenyek oxigénnel, vagy nedvességgel szemben. A reakció szelektivitása kitûnõ, emellett enyhe reakciókörülmények között, számos funkciós csoport (OH, NH, COOH, CHO) jelenlétében végbemegy anélkül, hogy szükség lenne védõcsoportok alkalmazására [30]. Mindezek a tulajdonságok elsõsorban az ón-szén kötés kis polaritásának köszönhetõk. A kapcsolást különösen eredményesen használják több funkciós-csoportot tartalmazó, bonyolult szerkezetû molekulák átalakítására. A Stille-kapcsolás hátránya viszont az ónvegyületek közismert toxikus hatása és a reakcióban keletkezõ trialkilón-halogenidek eltávolításának problémája
Ferrocénvázas vegyületek elõállításánál is találunk néhány olyan példát, ahol Stille-kapcsolást alkalmaztak, azonban ezekben az esetekben a ferrocénszármazék az ónorganikus reakciópartner (ferrocenil-tributil-sztannán) nem pedig a halogenid. A Stille-reakció tárgyalását ebben a fejezetben az teszi mégis indokolttá, hogy munkám során általam elõállított jód-ferrocén-származékok átalakítására alkalmaztam ezt a kapcsolási reakciót. (Lásd kísérleti rész 2.3.1 fejezet 2.6 egyenlet)
A Suzuki-reakció talán a keresztkapcsolási reakciók legsokoldalúbb fajtája. A felhasznált bórorganikus vegyületek szintén csökkent nukleofil karakterrel rendelkeznek (ezért számos funkcióscsoport jelenlétében kivitelezhetõ a kapcsolás védõcsoportok kialakítása nélkül is), valamint oxigénnel és nedvességgel szemben stabilak. A módszer további értékes tulajdonsága, hogy a kiindulási boronsavak és származékaik nem toxikusak, a reakció vizes közegben is elvégezhetõ és a képzõdõ bórsav nem zavarja a tisztítási eljárásokat. Fontos megemlíteni, hogy a ligandumcsere során - más keresztkapcsolási reakcióktól eltérõen - egy ekvivalens bázis jelenlétére van szükség. Mivel a szerves bórszármazék nukleofil ereje nem elég nagy, így az organopalládium-halogenidet kell elektrofilebbé tenni, a halogenidet alkoxi- vagy karbonátionra cserélve. Feltételezések szerint a reakcióközegben jelenlevõ alkoxid/karbonát/hidroxidionok nem csak a palládium elektrofil jellegét fokozzák, de a bórorganikus vegyület nukleofil jellegét is oly módon, hogy egyensúlyi addícióban anionos boronátkomplexet képeznek vele.
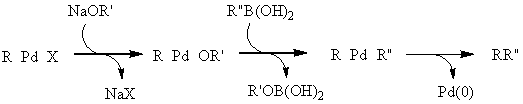
![]()
Aril-ferrocének elõállítására a Suzuki-kapcsoláson belül is több lehetõségünk van. Elõször is a ferrocenilcsoportot képviselheti egyrészt az aril-boronsav, másrészt a haloferrocén is. Mivel a ferrocén-boronsav és a jód-ferrocén (vagy bróm-ferrocén) elérhetõségét, stabilitását tekintve nem nagyon különböznek egymástól, a választást inkább a másik reakciópartner kezelhetõsége határozza meg. Döntõ jelentõségû a katalizátorrendszer, a bázis, illetve az oldószer megfelelõ megválasztása. Knapp és Rehahn összehasonlította a ferrocén-diboronsav / fenil-halogenid és dihaloferrocén / fenil-boronsav reakciópartner párok reakcióit két különbözõ rendszerben (A: toluol / 2M Na2CO3-oldat / Pd(PPh3)4; B: DME / 3M NaOH-oldat / PdCl2(dppf)). Az A módszer alkalmazásakor a dijód-ferrocén : fenil-boronsav pár esetén volt jóval nagyobb a konverzió (55% / 25%), míg a B esetén mindkét reakciópartner pár alkalmazása azonosan jó eredményt mutatott (konverzió: 90%) [42].
Imrie jód-ferrocént reagáltatott különféle szubsztituált fenil-boronsav származékokkal Suzuki-reakcióban. A paraméterek fokozatos változtatásával meghatározta az optimális reakciókörülményeket: 90%-os etanol mint oldószer, palládium-acetát katalizátor, és nátrium-karbonát, vagy bárium-hidroxid bázis bizonyult legelõnyösebbnek. [43]
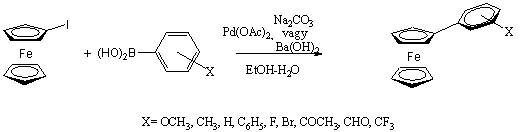
(1.15)
A benzol gyûrûhöz kapcsolódó elektronküldõ szubsztituensek növelik a reakció sebességét azáltal, hogy ezek a csoportok nukleofilabbá teszik a fenil-boronsav származékot. Elektronszívó szubsztituens esetén az ellenkezõje tapasztalható.
A keresztkapcsolási reakciók következõ változata, amelyet jód-, illetve dijód-ferrocén átalakítására is használtak, a cinkorganikus reagensek jelenlétében lejátszódó Negishi-reakció. A cinkorganikus vegyületek alkalmazása mellett szól, hogy viszonylag könnyen hozzáférhetõek, a funkciós csoportok egy jelentõs részénél nincs szükség védõcsoportok alkalmazására, és a cink és palládium közti ligandumcsere már szobahõmérsékleten is gyorsan lejátszódik. Hátrányuk, hogy nedvességre és oxigénre érzékenyek.
Jód-, illetve 1,1'dijód-ferrocén és egy cinkorganikus szerin származék palládium katalizált kapcsolási reakciójában az 1.16 egyenletben látható termékek keletkeztek. 1,1'-dijód-ferrocén kiindulási anyag esetén a cinkorganikus reagens feleslegének növelése a diszubsztituált ferrocén származék keletkezését jelentõs mértékben elõsegítette [44].
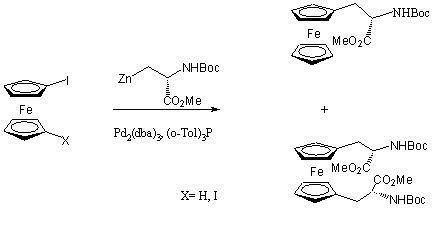
(1.16)
Aril-alkinek és konjugált eninek legkényelmesebb elõállítási módja az aril- vagy alkenil-halogenidek palládium katalizált kapcsolása terminális alkinekkel. Sonogashira nevéhez fûzõdik az a felismerés, hogy in situ elõállított alkinil-réz reagensek az elõbb említett kapcsolási reakcióba vihetõk.
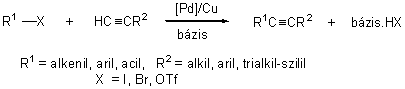 (1.17)
(1.17)
Ez a folyamat végsõ soron két katalitikus reakció: a palládiumkomplexek réz-katalizált alkinilezése (1.7 ábra "B" ciklus), valamint az sp2-szénatomokat tartalmazó halogenidek és terminális acetilének palládium jelenlétében lejátszódó kapcsolása kombinációjaként fogható fel (1.7 ábra "A" ciklus) [45]. Bár a reakció valószínûleg az 1.5.1.1. fejezetben bemutatott fõ lépéseket (oxidatív addíció, fémek közti ligandumcsere, reduktív elimináció) követve játszódik le, pontos mechanizmusa nem ismert. A katalitikusan aktív részecskék szerkezetével és a CuI kokatalizátor valódi szerepével kapcsolatban nem állnak rendelkezésünkre biztos adatok. Az oxidatív addíciót követõen a palládium(II)komplex reakcióba lép a terminális acetilénnel, valószínûleg egy átmenetileg képzõdõ réz-acetilid intermedier közremûködésével, majd a keletkezõ alkinil-palládium(II) vegyületbõl reduktív eliminációval képzõdik a termék. Katalizátorként leggyakrabban a PdCl2(PPh3)2 komplexet használják. Feltételezik, hogy a katalitikusan aktív részecske ekkor a Pd(II)komplex és az acetilén reakciójában képzõdõ Pd-acetilid komplexbõl jön létre reduktív elimináció útján (1.7 ábra "C" ciklus). Az elõbb említett palládiumvegyületen kívül általánosan alkalmazzák még a Pd(0)(PPh3)4, illetve Pd(0)2(dba)3 + foszfán típusú katalizátorokat is. A kis reakciókészségû aril-bromidok átalakításánál gyakran a 2/1-nél nagyobb foszfán/palládium aránnyal dolgoznak, hogy a szükséges magasabb hõmérsékleten elkerüljék a katalizátor bomlását, fémpalládium kiválását.

1.7 ábra A Sonogashira kapcsolás mechanizmusa
Mivel a kokatalizátorként használt réz-sók oxidatív körülmények között elõsegítik az alkinek úgynevezett Glaser-típusú homokapcsolását, mely a megfelelõ szimmetrikus diint eredményezi melléktermékként, az utóbbi néhány évben számos kutatócsoport koncentrált a réz-mentes Sonogashira kapcsolás megvalósítására [46-49].
Ferrocénvázas vegyületek esetében a jód-ferrocén, illetve a dijód-ferrocén és a legkülönbözõbb terminális alkinek (fenil-acetilén, acetilén [50], 1,4-dietinil-benzol [51, 52], 4-etinil-piridin, 5-etinil-bipiridin [53]) kapcsolása, változatos ferrocénvázas acetilén származékok képzõdését eredményezte 45-96%-os hozamok mellett.
A s-kötésû átmenetifém-organikus vegyületekbõl kiinduló szén-szén kötés kialakulásához vezetõ folyamatok közül a keresztkapcsolási reakciók mellett szintén nagy jelentõségû az alkén karbopalladálásából, majd az azt követõ b-eliminációból álló folyamat, amelyet felfedezõjérõl Heck-reakciónak neveztek el. A 1.8 ábrán látható katalitikus ciklus elsõ lépése ebben az esetben is az aril-halogenid és a palládium között lejátszódó oxidatív addíció. A kialakult σ-kötésû palládiumorganikus vegyülethez koordinálódik az alkén, majd a beékelõdést követõen olyan alkilpalládium-komplexhez jutunk, melybõl egy rotáció után kialakul az eliminációhoz szükséges szin-periplanáris Pd-C-C-H elrendezõdés. Így lehetõség nyílik a β-elimináción keresztül megvalósuló stabilizációra, mely az alkén termék képzõdéséhez vezet. A hidrogén-halogenid eliminációja, mely a hozzáadott bázis hatására megy végbe, a katalitikus ciklus záródását, az aktív katalizátor regenerálását eredményezi.
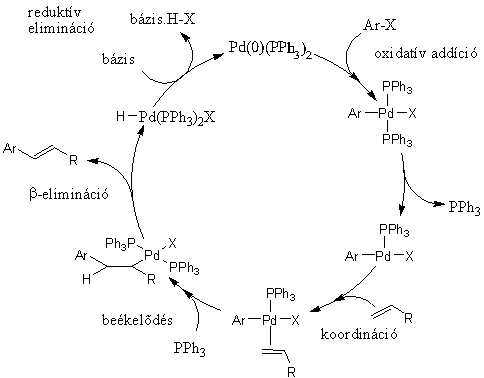
1.8. ábra A Heck-reakció mechanizmusa
Olyan Heck-reakciókban, amikor a β-elimináció során nem az eredeti C=C kötést regeneráljuk, a reakcióban általában új kiralitáscentrum alakul ki. Ilyen esetekben királis ligandumok alkalmazásával elvileg lehetõségünk van enantioszelektív átalakításokra.
A Heck reakció során elérhetõ konverzió és szelektivitás természetesen jelentõs mértékben függ a reakciópartnerektõl, de emellett befolyásolható a körülmények - oldószer, katalizátor, bázis, esetleg valamilyen egyéb adalék - megfelelõ megválasztásával. Az eredetileg Heck által közölt Pd(OAc)2/PPh3/Et3N katalizátor-bázis párosításon kívül általánosan alkalmazzák a Jeffery [54] által kidolgozott Pd(OAc)2/K2CO3/nBu4NCl rendszert DMF vagy DMSO oldószerben. Ez utóbbi esetben a kvaterner ammóniumsó szerepe a feltételezések szerint az, hogy a kialakuló palládiumkolloidot stabilizálja, megakadályozza az aggregációt [55]. E két, már-már klasszikussá váló módszer mellett számos katalizátor-bázis-oldószer összeállítást kipróbáltak már.
Kasahara és munkatársai írták le elõször olefinek palládium katalizált reakcióit jód-, illetve 1,1'-dijód-ferrocénnel. A szokásos Heck körülmények között reagáltatták a halogeno-ferrocéneket az alkének legkülönbözõbb képviselõivel: egyszerû olefinekkel (sztirol), akrilsav származékokkal (etil-akrilát, metil-metakrilát, akrilnitril), telítetlen ketonokkal (metil-vinil-keton, fenil-vinil-keton), allil-alkoholokkal (allil-alkohol, 3-butén-2-ol) és telítetlen acetátokkal (vinil-acetát, izopropenil-acetát, acetofenon-enol-acetát).
(1.18)
Allil-alkoholok és jód-ferrocén reakciója minden esetben ferrocén-karboxaldehidet vagy -ketont eredményezett.
(1.19)
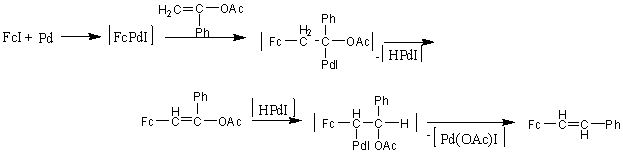
Vinil- és izopropil-acetát jód-ferrocénnel való Heck-kapcsolása diferrocenil-etilén, illetve 1,2-diferrocenil-1-propén termékhez vezetett, míg acetofenon-enol-acetát esetében sztiril-ferrocénbõl és 1-acetoxi-1-fenil-2-ferrocenil-etilénbõl álló termékelegyet kaptak. A sztiril-ferrocén képzõdése a következõképpen játszódhat le: a ferrocenilcsoportot b-pozícióban tartalmazó terméket eredményezõ b-eliminációt követõen a hidrido-palládium komplex újra addícionálódik oly módon, hogy most palládium a ferrocénnel szomszédos szénatomra kerül. Egy Pd(OAc)I komplex eliminációt követõen megkapjuk a sztiril-ferrocént [50].
(1.20)
Frejd és Calström 1,1'-dijód-ferrocént reagáltatott Heck reakcióban etil-2-[(terc-butoxi-karbonil)-amino]-akriláttal Jeffery körülmények között (1.21 egyenlet) [56].
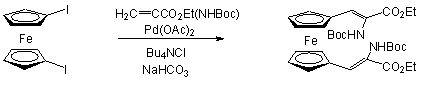
A regioszelektivitást befolyásolja az olefin molekula kettõs kötésén lévõ szubsztituens donor/akceptor tulajdonsága: elektron-donor szubsztituens (pl. alkoxicsoport) jelenléte az a-helyzetben helyettesített termék mennyiségét növeli [57]. Herrmann és munkatársai ferrocenil-etenil-szilatrán szintézisét valósította meg jód-ferrocén és 1-etenil-szilatrán Heck-kapcsolásával. Mivel a szilatráncsoport erõs elektron-donornak mondható, a fent említettek alapján a kapcsolás eredményeként a- és b-pozícióban helyettesített termékek elegye várható. Meglepetésre azonban kizárólag a-termék keletkezett, függetlenül attól, hogy milyen körülmények között (katalizátor, oldószer, bázis, stb.) történt a reakció. Ezek változtatása ugyanis csak a konverziót befolyásolta, a regioszelektivitásra nem volt hatással [58].
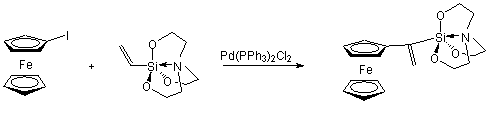 (1.22)
(1.22)
Az a-termék kizárólagos keletkezése a szilatráncsoport elektrondonor sajátságán túlmenõen valószínûleg annak köszönhetõ, hogy a nagy térkitöltésû csoport miatt sztérikus gátlás lép fel. Az 1.8 ábrán bemutatott beékelõdési lépésben a szintén nagy térigényû palládium-komplex inkább a b-helyzetû szénatomra kerül.
Mivel a palládiumkomplexek alkalmasak a szén-monoxid aktiválására, palládiumkatalizátorok segítségével ezt a molekulát közvetlenül beépíthetjük szerves vegyületekbe. A fémorganikus reakciópartnerekkel, vagy alkinekkel végbemenõ kapcsolási reakciók módosításával, inert körülmények helyett szén-monoxid atmoszféra alkalmazásával különbözõ karbonilvegyületek szintézisére nyílik lehetõség. Egyéb nukleofil reakciópartnerek (víz, alkoholok, aminok) jelenlétében karbonsavszármazékokhoz juthatunk.
A szén-monoxid atmoszférában lejátszódó reakciók mechanizmusa nagyon hasonló az eddig ismertetett, palládiumkomplexek jelenlétében lejátszódó folyamatokéhoz. Az egyetlen lényeges különbség, hogy a katalitikusan aktív Pd(0)komplex és az aril/alkenilhalogenid, vagy -triflát reakciójában képzõdõ aril/alkenil-palládium(II) vegyületbõl a szén-monoxid beékelõdése révén acil-palládium(II) komplex alakul ki. Ez a továbbiakban reakcióba léphet egy fémorganikus reagenssel (1.10 ábra, "A" reakcióút), acetilén-származékokkal ("B" reakcióút),

1.9 ábra A szén-monoxid atmoszférában lejátszódó palládium-katalizált reakciók mechanizmusa
vagy nukleofil vegyületekkel ("C" reakcióút). Az elsõ reakciótípusnál az 1.5.1.1. fejezetben leírtakhoz hasonlóan a fémek közti ligandumcsere vezet a termék kialakulásához és a katalitikusan aktív részecske regeneráláshoz. "A", "B" és "C" reakcióút esetén a termék képzõdését HPd(II)L2X típusú komplex kialakulása kíséri, melybõl bázis jelenlétében HX reduktív eliminációja révén nyerjük vissza a Pd(0)komplexet.
A karbonilezés során alkalmazott katalizátorok, bázisok és oldószerek alapvetõen megegyeznek az inert atmoszférában lejátszódó kapcsolási reakcióknál ismertetettekkel.
Szerves-halogenidek palládium-katalalizált karbonilezése nukleofil reakciópartner jelenlétében - mely karbonsavakat, észtereket, karbonsav-amidokat, vagy egyéb karbonsav származékokat eredményez - széles körben alkalmazott folyamat mind a laboratóriumi szintézisekben, mind pedig az ipari eljárásokban.
Az aminokarbonilezési reakció mechanizmusa több módon képzelhetõ el (1.10. ábra). A palládium-katalizált homogénkatalitikus reakciók régóta tanulmányozott mechanizmusa alapján elfogadott tény az organopalládium-halogenid kialakulása a szerves-halogenid katalitikusan aktív Pd(0) komplexre történõ oxidatív addíciója révén. A katalitikus reakció további lefolyása azonban erõsen függhet a reakció körülményeitõl.
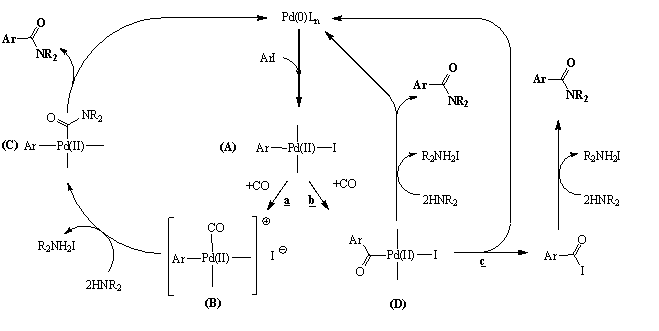
1.10. ábra A palládium-katalizált aminokarbonilezés reakciómechanizmusa
Az egyik alternatíva (1.10. ábra, "a" reakcióút), hogy az aril-halogenid oxidatív addícióját követõen kialakult komplexhez (A) egy szén-monoxid molekula koordinálódik és kialakul egy aril-karbonil-palládium-komplex (B). Ebbõl bázis jelenlétében az amin támadása révén kialakul egy aril-karbamoil-palládium komplex (C). Végül egy reduktív eliminációs lépés segítségével jutunk az ArCONR2 termékhez [59, 60].
A másik lehetõség, amikor egy szén-monoxid molekula beékelõdésével egy acil-palládium komplex (D) keletkezik. Ez reakcióba lép a nukleofil reagenssel (HNR2), melynek hatására felszabadul az ArCONR2 termék és egy bázis hozzáadásával visszakapjuk a katalitikusan aktív Pd(0) komplexet (1.10 ábra "b" reakcióút) [61].
Egyes feltételezések szerint nem zárható ki az sem, hogy az acil-palládium komplexbõl (D) eliminációval savhalogenid képzõdik, amely egyszerû SN reakcióban acilezi az amint (1.10. ábra "c" reakcióút) [62].
Valószínû, hogy a kiindulási vegyületek tulajdonságaitól függõen egyik, vagy másik, vagy akár - különbözõ sebességgel - mindhárom reakciósor lejátszódik [59]. Hogy melyik mechanizmus milyen mértékben valósul meg, az függhet a reakciókörülményektõl (pl. az oldószertõl, az amin bázicitásától és sztérikus sajátságától, az aril-, vagy alkenil-halogenid tulajdoságától, az alkalmazott ligandum természetétõl, stb.).
Aril-halogenidek homogénkatalitikus karbonilezési reakciói során végbemehet kettõs karbonilezés is. Ekkor a nukleofil reakciópartnertõl függõen különféle a-keto-karbonsavszármazékok keletkeznek. Az aril-halogenidek ilyen típusú átalakítását a-keto-amidokká 1982-ben egymástól függetlenül fedezte fel Yamamoto és Ozawa [63], illetve Tanaka és Kobayashi [64].
Ezt követõen részletes kutatásokat végeztek a kettõs karbonilezés mechanizmusának felderítésére. A reakció alapvetõ lépéseit az 1.11 ábra mutatja be: az 1. lépés az oxidatív addíció, a 2. lépés a CO beékelõdés. A 3. lépést, mely a szekunder amin reakciója az acil-palládium komplexszel (D) szén-monoxid jelenlétében, kétféleképpen képzelték el [65].
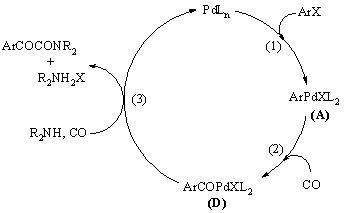
1.11 ábra Aril-halogenidek katalitikus kettõs karbonilezésének elemi lépései szekunder amin jelenlétében.
Az elsõ feltételezések szerint az elsõ CO beékelõdést egy újabb CO inzerció követheti. Ez egy glioxil-palládium komplexet (E) eredményez, melybõl az amin támadás révén jutunk az a-keto-amid termékhez (1.23 egyenlet). Részletes vizsgálatok azonban cáfolták ezt az elképzelést.
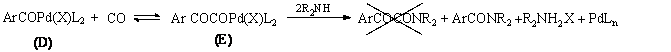
Yamamoto és munkatársai elõzetesen kipreparált glioxil-palládium komplexet (E) reagáltattak szekunder aminnal, melynek eredményeként nem a várt ketoamid, hanem az amid keletkezett [66].
A másik eshetõség szerint az elsõ CO beékelõdését egy másik koordinációja követi, ezzel egy acil-karbonil-palládium komplexhez (F) jutunk, melybõl az amin támadása révén egy acil-karbamoil-palládium komplexet (G) kapunk. Végül az a-keto-amid reduktív eliminációval képzõdik a (G) komplexbõl (1.24 egyenlet).

A kettõs karbonilezést számos tényezõ befolyásolja. Ilyen lehet a nukleofil reagensként használt amin természete (sztérikus és elektronikus sajátságai), a felhasznált ligandum, a szubsztrátum tulajdonságai, a szén-monoxid nyomása, a reakció hõmérséklete, a hozzáadott bázis és az oldószer minõsége.
A legjobb szubsztrátumnak az aril-jodidok bizonyultak. A para helyzetben elektronszívó csoporttal (CN, CF3) helyettesített aril-halogenid reakciókészsége megnõ a nem szubsztituált származékhoz képest, ugyanakkor az a-ketoamid képzõdés szelektivitása lecsökken. Elektronküldõ csoport jelenléte esetén az ellenkezõje tapasztalható.
A CO nyomás növelése kedvez az a-ketoamid kialakulásának, azonban a túl nagy nyomás jelentõsen csökkentheti a reakció sebességét.
A bázikusabb szekunder aminok hatékonyan alkalmazhatóak a kettõs karbonilezési reakciókban, de az aminok geometriája jelentõsen befolyásolja mind a szelektivitást, mind pedig a konverziót. Például az elágazó alkilcsoportokkal rendelkezõ di-izo-butil-amin teljesen inaktívnak bizonyult és a di-izo-propil-amin jelenlétében is csak amid képzõdést és csekély átalakulást figyeltek meg. A kisebb térkitöltésû csoportokkal rendelkezõ szekunder aminok azonban eredményesen alkalmazhatóak karbonilezési reakcióban, eltérõ szelektivitással [59].
Watanabe és
munkatársai a hozzáadott bázis, illetve az oldószer hatásait vizsgálták di(m-kloro)bisz-(h -allil)dipalládium(II)
és trifenil-foszfán katalizátor rendszer alkalmazásával ( P/Pd = 2/1 ), 25°C-on,
légköri CO nyomáson. A különbözõ bázisok markáns különbségeket okoztak a ketoamid
hozamát tekintve. A Watanabe által vizsgált bázisok közül
(1,4-diazabiciklo[2.2.2]oktán (DABCO), trietil-amin, piridin,
1,8-diazabiciklo[5.4.0]undec-7-én (DBU)), a DABCO bizonyult messze a
leghatékonyabbnak tetrahidrofurán oldószerben [67]. Érdekes, hogy a DBU ilyen
körülmények között a-ketoamid keletkezésére
nézve teljesen hatástalan volt, míg az Inoue által kipróbált rendszerben (
Láthatjuk, hogy a különbözõ kutatócsoportok vizsgálatai alapján más-más reakciókörülmények adódnak optimálisnak, a reakció mechanizmusát tekintve azonban nagyjából egyetértés van (1.12 ábra). A különbözõ bázisok okozta eltérések magyarázata még nem teljesen tisztázott.

1.12 ábra A kettõs karbonilezési reakció mechanizmusa.
A Pannon Egyetem Szerves Kémia tanszékén évtizedes múltra tekint vissza alkenil- és aril-halogenidek és -triflátok homogénkatalitikus karbonilezési reakcióinak vizsgálata [69-71]. Ezen kutatások keretein belül irányult a figyelem a jód-ferrocén kiindulási anyagként való felhasználására aminokarbonilezési reakcióban. Jód-ferrocént reagáltattak különbözõ szekunder aminokkal Pd-katalizátor jelenlétében szén-monoxid nyomás alatt. A kísérletek eredményeként új típusú ferrocénvázas a-ketoamidokat szintetizáltak, melléktermékként ferrocénkarbonsavamidok képzõdtek [72].
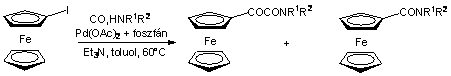
(1.25)
Megállapították, hogy a reakció szelektivitása jelentõs mértékben függ a hõmérséklettõl. Alacsonyabb hõmérsékleten a legtöbb esetben az a-ketoamidok voltak a fõ termékek, míg a hõmérséklet növelése a monokarbonilezésnek kedvezett. A nyomás növelése egy ideig a konverzió és az a-ketoamidok hozamának növekedéséhez vezetett, ugyanakkor a nyomás 40 bar fölötti növelése az átalakulás csekély mértékû visszaesését eredményezte, míg az amid/ketoamid arány nem változott. A jelenséget azzal magyarázták, hogy a magas CO nyomás hatására megnövekszik a CO ligandumok száma a Pd koordinációs övezetében, ami nehezíti a jód-ferrocén oxidatív addícióját, ezáltal csökken a reakció sebessége.
A karbonilezés szelektivitását nagy mértékben befolyásolta a nukleofil reagensként alkalmazott amin térigénye. Míg a kisebb térkitöltésû szekunder aminok esetén (morfolin, Et2NH, nBuNH, piperidin, 3,5-dimetil-piperidin) az a-ketoamidot sikerült 60 oC-on jó hozammal elõállítani, addig a terjedelmesebb, szerteágazóbb alkilcsoporttal rendelkezõ szekunder aminoknál (cHex2NH, 2-etil-piperidin, 2,6-dimetil-piperidin) - feltehetõen az amin sztérikus gátlásának köszönhetõen - a ketoamid keletkezése tekintélyes mértékben háttérbe szorult.
Az elõbb leírt megfigyeléseket követõen kapcsolódtam be a kutatásba. Elsõként azt vizsgáltam, hogy a jód-ferrocén aminokarbonilezési reakciójában alkalmazhatók-e aminosav-észterek mint nukleofil reagensek. Majd az 1,1'-dijód-ferrocén hasonló típusú átalakítását tanulmányoztam mind aminok, mind aminosav-észterek jelenlétében, különös tekintettel az 1,1'-heterodiszubsztituált származékok elõállítására.
Kísérleteim során elõször a jód-ferrocén (1) és glicin-metil-észter reakcióját vizsgáltam (2.1. egyenlet).

(2.1.)
A reakciókörülmények kiválasztásánál a korábbi karbonilezési eredményeket vettem alapul (ld. 1.5.2.2. fejezet): az optimálisnak talált 40 bar CO nyomáson dolgoztam, katalizátor prekurzorként a korábbi jód-ferrocén karbonilezési kísérletekhez hasonlóan Pd(OAc)2 és 2 ekvivalens PPh3 elegyét használtam.
A katalitikus hatást ekkor egy Pd0 komplex fejti ki, amely in situ keletkezik a Pd(OAc)2 -ból a foszfin jelenlétében [73]. A PPh3 ezenkívül ligandumként is szolgál.
 (2.2.)
(2.2.)
Az alkalmazott katalizátor óriási elõnye az egyéb palládium-tartalmú katalizátorrendszerekkel szemben, hogy sem a Pd-só, sem a ligandum nem igényel különleges tárolási feltételeket, nem érzékeny oxigénre és a levegõ nedvességtartalmára, továbbá viszonylag olcsó.
Az elõkísérletek során megállapítottam, hogy a szabad aminosav mint nukleofil nem alkalmazható a karbonilezés során. Ezért a továbbiakban a könnyen elõállítható metil-észterek reakcióit vizsgáltam. A reakciók elõrehaladását vékonyréteg-kromatográfiával, illetve gázkromatográfiás módszerrel követtem.
A glicin-metil-észter (Gly-OMe) mint nukleofil reakciópartner esetében kapott eredményeket az 1. táblázat mutatja be. A korábbi karbonilezési kísérletek alapján a jód-ferrocén (1) karbonilezése amid (2a), illetve a-ketoamid (3a) típusú termékekhez vezethet.
Toluol oldószerben trietil-amin bázis jelenlétében bár teljes átalakulást tapasztaltam, a gázkromatográfiás elemzés szerint a 2a amidszármazék csak 96 %-os hozammal keletkezett. A 3a ketoamid képzõdését nem tudtam kimutatni. Mivel azt tapasztaltam, hogy az aminosav-észter hidroklorid rosszul oldódik toluolban, ezért a továbbiakban THF-ben dolgoztam. Ekkor 100 oC-on teljes átalakulással, jó szelektivitással játszódott le a karbonilezés.
1. táblázat Jód-ferrocén (1) karbonilezése glicin-metil-észter jelenlétébena
|
Sorsz. |
Bázis |
Bázis/1 (mol/mol) |
Oldószer |
Hõm. (oC) |
R.idõ (h) |
Konv.b (%) |
Hozamb (%) |
|
|
2a |
3a |
|||||||
|
1 |
Et3N |
6 |
toluol |
100 |
8 |
100 |
96 |
0 |
|
2 |
Et3N |
6 |
THF |
100 |
8 |
100 |
100 |
0 |
|
3 |
Et3N |
6 |
toluol |
60 |
8 |
71 |
58 |
5 |
|
4 |
Et3N |
6 |
THF |
60 |
8 |
82 |
73 |
9 |
|
5 |
DBU |
6 |
THF |
100 |
8 |
100 |
58 |
4 |
|
6 |
DBU |
8 |
THF |
100 |
8 |
96 |
9 |
32 |
|
7 |
DBU |
8 |
THF |
60 |
12 |
100 |
8 |
13 |
|
8 |
DBU |
10 |
THF |
100 |
2 |
100 |
4 |
12 |
aReakciókörülmény: jód-ferrocén (1) (0,5 mmol), glicin-metil-észter hidroklorid (2,5 mmol), Pd(OAc)2 (0,025 mmol), PPh3 (0,05 mmol), 12,5 ml oldószer, 40 bar CO. bGC alapján (belsõ standard: eikozán).
Mivel a korábbi eredmények alapján a hõmérséklet csökkentése az a-ketoamidok képzõdésének kedvez [72], további kísérleteket végeztem a 3a vegyület elõállítása érdekében.
Trietil-amin bázis jelenlétében lejátszódó reakcióban 60°C-on ugyan mind toluol, mind THF oldószerben sikerült 3a képzõdését kimutatnom, de a fõ termék továbbra is az amid (2a) maradt.
Mivel úgy tûnt, hogy a hõmérséklet csökkentésével döntõen nem befolyásolható a reakció szelektivitása, a további kísérletekhez az irodalom alapján egy másik bázist, a DBU-t választottam. Inoue és munkatársai jód-benzol dietil-amin jelenlétében lejátszódó karbonilezésénél ugyanis megfigyelték, hogy míg piridin, vagy dietil-amin bázis hozzáadásával közepes szelektivitást értek el, addig DBU jelenlétében jó hozammal keletkezett a megfelelõ a-ketoamid. [68]
Mivel a nukleofil reakciópartner, a glicin-metil-észter hidroklorid a szubsztrátumhoz képest ötszörös feleslegben volt jelen, a jód-ferrocénre nézve 6, majd 8, végül 10 ekvivalens mennyiségû DBU-val dolgoztam.
Míg 6 ekvivalens DBU esetén az a-ketoamid/amid arány a korábbi, 60 oC-on végzett kísérletekhez képest nem növekedett, a DBU mennyiségét 8 ekvivalensre emelve e tekintetben jelentõs javulást figyeltem meg. A termékek hozama azonban a gázkromatográfiás eredmények alapján nagyon alacsony volt.
A termékelegy részletesebb vizsgálata azt mutatta, hogy ez többféle okra vezethetõ vissza. Egyrészt a vékonyréteg-kromatográfiás vizsgálatok szerint a termékek DBU jelenlétében nem inert körülmények között már szobahõmérsékleten is bomlást szenvedtek. Megfigyeltem viszont, hogy 2a és 3a THF-es oldata órákon át stabil maradt, így a továbbiakban a feldolgozás elsõ lépését, a DBU eltávolítását inert körülmények között végeztem, így a bomlást sikerült elkerülnöm (ld. 2. táblázat).
DBU jelenlétében a két termék a gázkromatográfiás vizsgálat körülményei között is bomlott, a bázis mennyiségének növelésével párhuzamosan csökkenõ hozamot részben ez is okozta (ld. 1. táblázat 5-8. sor). Meg kell jegyeznem, hogy a kiindulási vegyület, a jód-ferrocén DBU jelenlétében is stabil maradt.
A 2a és 3a termékek alacsony hozamát azonban nem csupán a DBU jelenlétében tapasztalható bomlás okozta. A vékonyréteg-kromatográfiás elemzés 2a és 3a mellett még további két, egy sárga és egy piros színû termék képzõdését is mutatta.
A melléktermékeket
oszlopkromatográfiás elválasztás után különbözõ analitikai vizsgálatoknak (1H-
és 13C-NMR, valamint 1H- 1H és 1H-
Az NMR spektrumok két nagyon
hasonló szerkezetû ferrocénvázas vegyület képzõdését mutatták. Az 1H-NMR
spektrumokban a ferrocénvázhoz tartozó jeleken kívül az 1,4-3,6 ppm
tartományban összesen 16 protonnak megfelelõ több multiplett, valamint a piros
vegyület esetében 8,00; a sárga vegyületnél pedig 7,38 ppm-nél egy széles
szingulett található. Az 1H-
A 13C-NMR spektrumok a 8 metilén-jelen és a ferrocénvázhoz tartozó jeleken kívül a piros terméknél 3 (190,9; 176,5 és 162,1 ppm-nél), a sárga terméknél két további jelet (177,2 ppm-nél és 170,2 ppm-nél) tartalmaznak. Az aminok, mint nukleofil reagensek jelenlétében korábban a tanszéken elõállított amidok és ketoamidok spektrumait alapul véve [72], a sárga színû vegyület esetén a 170,2 ppm-nél jelentkezõ jel a ferrocénvázas amidok C=O eltolódásával, míg a piros színû termék esetén 162,1 ppm-es jel a ferrocénvázas ketoamidok amidocsoportjának, 190,9 ppm-nél pedig ketocsoportjának C=O eltolódásával egyezik meg.
Mindezek alapján feltételezhetjük, hogy a két vegyület a ferrocén és DBU reakciójából származó amid-, illetve ketoamid típusú termék.
Az irodalmi adatok alapján a DBU dimetil-karbonát [74], vagy dibenzil-karbonát [75] jelenlétében acilezhetõ, illetve alkil-halogenidekkel alkilezhetõ [76]. A termékek a megfelelõ sók. Az alkilezésnél vízzel lejátszódó reakcióban egy, a két gyûrût összekötõ híd felnyílásával képzõdõ terméket is kimutattak. Leírtak egy ruténium-komplex jelenlétében lejátszódó acilezési reakciót is, melyben az elsõdlegesen képzõdõ só hidroxidion hatására alakul át a végtermékké [77].

2.1. ábra A jód-ferrocén és a DBU karbonilezési körülmények között lejátszódó reakciójának feltételezett lépései
A jód-ferrocén és DBU reakciójában az irodalomban leírt acilezési reakciók alapján a 4a,b só képzõdését feltételezhetjük, amely utána 5a,b vagy 6a,b termékekké bomolhat, a Ru-komplex jelenlétében lejátszódó, illetve az alkilezési reakcióban tapasztaltak analógiájára.
Az NMR spektrumok a 6a,b termékek képzõdését valószínûsítik. Ekkor a 8,00; illetve 7,38 ppm-nél található jelek az NH protonok jelei, melyek az 1H-1H korrelációs spektrumok alapján valóban csatolnak a szomszédos metilén-protonokkal. Az NH protonok nagy eltolódása valószínûleg azzal magyarázható, hogy az eredetileg kétgyûrûs vegyület hídja ugyan felnyílik, de hidrogénhíd kötés alakul ki a karbonilcsoport és az aminocsoport H-atomja között.
A termékek szerkezetét a vegyületekrõl készített tömegspektrumok is bizonyítják, a molekulaion tömege 6a esetében 382-nek, míg 6b-nél 410-nek adódott.
Érdekes megjegyezni, hogy a jód-ferrocén aminokarbonilezése DBU mint nukleofil reagens jelenlétében 6a képzõdését eredményezte jó szelektivitással, 6b pedig csak nyomokban volt jelen a reakcióelegyben. Ezzel ellentétben a glicin-metil-észter és jód-ferrocén reakciójában DBU jelenlétében kapott melléktermékek fõ alkotóeleme 6b volt.
A fenti eredmények alapján elvégeztem a jód-ferrocén karbonilezését glicin-metil-észter (2.1. egyenlet) és négy további aminosav-észter (2.3. egyenlet) jelenlétében az 1. táblázat 2. valamint 6. sorának megfelelõ reakciókörülmények között. A DBU bázis hozzáadásával kivitelezett reakciók esetén a termékelegy feldolgozását inert körülmények között is végrehajtottam.

2. táblázat Jód-ferrocén (1) karbonilezése különféle aminosav-észterek jelenlétébena
|
Sorsz. |
Nukleofil reagens |
Bázis |
Konv.b (%) |
Izolált hozam (%) |
||
|
2 |
3 |
6a+6b |
||||
|
1 |
Gly-OMe |
Et3N c |
100 |
94 |
- |
- |
|
2 |
Gly-OMe |
DBU d |
99 |
1 |
25 |
2 |
|
3 |
Gly-OMe |
DBU e |
99 |
1 |
89 |
5 |
|
4 |
L-Ala-OMe |
Et3N c |
92 |
87 |
- |
- |
|
5 |
L-Ala-OMe |
DBU d |
99 |
17 |
20 |
14 |
|
6 |
L-Ala-OMe |
DBU e |
99 |
23 |
28 |
21 |
|
7 |
L-Phe-OMe |
Et3N c |
85 |
81 |
- |
- |
|
8 |
L-Phe-OMe |
DBU d |
97 |
2 |
10 |
29 |
|
9 |
L-Phe-OMe |
DBU e |
96 |
15 |
26 |
42 |
|
10 |
L-Met-OMe |
Et3N c |
81 |
75 |
- |
- |
|
11 |
L-Met-OMe |
DBU d |
98 |
3 |
17 |
19 |
|
12 |
L-Met-OMe |
DBU e |
98 |
4 |
37 |
32 |
|
13 |
L-Pro-OMe |
Et3N c |
90 |
84 |
- |
- |
|
14 |
L-Pro-OMe |
DBU d |
100 |
32 |
29 |
3 |
|
15 |
L-Pro-OMe |
DBU e |
100 |
32 |
30 |
19 |
aReakciókörülmény: jód-ferrocén (0,5 mmol), aminosav-metil-észter hidroklorid (2,5 mmol), Pd(OAc)2 (0,025 mmol), PPh3 (0,05 mmol), 12,5 ml THF, 40 bar CO, 8h bGC alapján (belsõ standard: eikozán). c Et3N/1=6. d DBU/1=8. e DBU/1=8, a tisztítás során a DBU eltávolítása inert körülmények között történt.
A Et3N bázis jelenlétében lejátszódó reakciókban
DBU jelenlétében változó mennyiségû 6a+6b terméket tudtam elkülöníteni. E kísérletek során csak ezek együttes mennyiségét határoztam meg, mert elválasztásuk csak egy további oszlopkromatográfiás eljárással volt lehetséges.
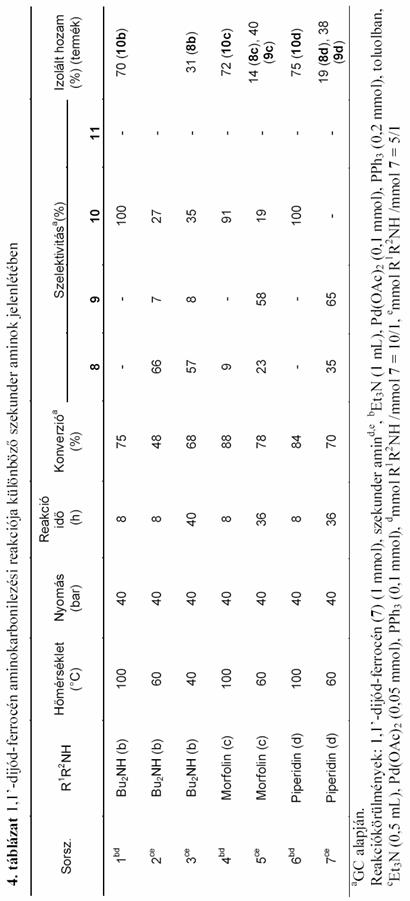
A jód-ferrocén aminokarbonilezési reakciójához hasonlóan vizsgáltam az 1,1'-dijód-ferrocén (7) karbonilezését különbözõ szekunder aminok jelenlétében. Ez a reakció a korábbihoz képest bonyolultabb, hiszen míg az elõzõ esetben kétféle termék keletkezésével kellett számolni, addig 7 aminokarbonilezése elvileg ötféle vegyület (2.4 egyenlet 8-12) képzõdéséhez vezethet.

Az 1,1'-dijód-ferrocént 100°C-on, 40bar CO nyomáson az in situ Pd(OAc)2 + 2 PPh3 katalizátor jelenlétében reagáltattam különbözõ szekunder aminokkal. A korábbi jód-ferrocén aminokarbonilezési vizsgálatok alapján [72] azt vártam, hogy a ketoamid típusú termékek (9, 11, és 12) ezen a hõmérsékleten nem, vagy kis mennyiségben keletkeznek. Kettõs karbonilezés lejátszódását valóban nem tudtam kimutatni, de dietil-amin, dibutil-amin és piperidin alkalmazása esetén (3. táblázat 1; 4. táblázat 1, 3, 8 sor) meglepetésemre a dijód-ferrocén részleges átalakulása mellett is csak szelektíven a megfelelõ ferrocén-1,1'-dikarboxamid (10a, 10b és 10d) keletkezett, 8 jelenlétét nem tudtam kimutatni. Ez azt jelenti, hogy 100°C-on a második jód szubsztitúciója gyorsabb az elsõnél, ezáltal a 8 vegyület nem tud feldúsulni a termékelegyben, mert rögtön átalakul a 10 diamiddá. Különösen szembetûnõ ez a dibutil-amin esetén (4. táblázat 1. sor) ahol 8 óra elteltével a dijód-ferrocén 75%-os átalakulása mellett kizárólag az 1,1'-N,N-dibutil-ferrocéndikarboxamid (10b) jelenlétét tudtam a reakcióelegyben kimutatni. Morfolin nukleofil reagensként való használatakor 8 óra elteltével a ferrocén-dikarboxamid (10c) mellett, mintegy 1/10 részben 1'-jód-morfolino-ferrocénkarboxamid (8c) is jelen volt (4. táblázat 4.sor).
Ezek után azt vizsgáltam, hogy a hõmérséklet csökkentése milyen változásokat okoz a termékelegy összetételében. Az alacsonyabb hõmérsékleten kivitelezett reakció, a vélhetõ reakciósebesség csökkenések miatt, az 1,1'-dijód-ferrocén részleges szubsztitúciójából eredõ termékek (8, 9) arányának növekedését vetíti elõre, illetve a korábbi jód-ferrocén aminokarbonilezési tapasztalatok alapján, kettõs karbonilezett termékek (9, 11, 12) megjelenése is várható. Az esetlegesen így kapott termékek egyrészt újszerûek, másrészt 8, illetve 9 1'-jód-ferrocén származékok feltételezhetõen jó kiindulási anyagai lehetnek egy újabb aminokarbonilezési lépésnek, vagy egyéb heterodiszubsztituált ferrocénszármazékokat eredményezõ homogénkatalitikus kapcsolásnak. Ezen megfontolások mentén haladva az 1,1'-dijód-ferrocén dietil-amin jelenlétében lejátszódó karbonilezési reakciót vettem górcsõ alá (3. táblázat).
A hõmérséklet csökkentéssel nem titkolt célom volt, hogy 1'-jód-ferrocén származékokat minél nagyobb mennyiségben szintetizáljak. Mivel így a helyettesítendõ jódatomok mennyisége felére csökkent, a katalizátor és a bázis mennyiségét is felére csökkentettem.
A 60°C-on elvégzett kísérlet eredménye igazolta várakozásom helyességét. Nyolc órás reakcióidõt követõen ugyan még mindig az 1,1'-N,N-dietil-ferrocéndikarboxamid (10a) dominált, de mellette további három termék (8a, 9a, 11a) jelenlétét is ki tudtam mutatni a termékelegy vizsgálata során (3. táblázat 2. sor). 1,1'-N,N-dietil-ferrocéndiglioxilamid (12a) keletkezését még nyomokban sem tapasztaltam. A kísérletet 50 bar CO nyomáson megismételve 8a és 9a termékek arányának további növekedése figyelhetõ meg (3. táblázat 3.sor). Ez egyrészt írható a magasabb CO nyomás okozta reakciósebesség csökkenés számlájára (1.5.2.2. fejezet), másrészt a magasabb CO koncentrációból eredõen nõhet 9a kettõs karbonilezett termék mennyisége. 12a keletkezését továbbra sem tudtam kimutatni. Itt jegyzem meg, hogy a 12 vegyület keletkezését semmilyen más általam kipróbált körülmény, illetve nukleofil reagens esetén sem tapasztaltam.

Visszatérve az általánosan használt 40 bar CO nyomásra tovább csökkentettem a reakció hõmérsékletét. 40°C-on a 8a 1'-jód-ferrocénkarboxamid már tekintélyes arányban van jelen a termékelegyben (3. táblázat 5-10sor), igaz a reakcióidõ elõrehaladtával ez az arány számottevõen csökken az 1,1'-ferrocén-dikarboxamid keletkezésének köszönhetõen, de mennyiségét tekintve a 16-ik óráig növekedés tapasztalható, ahol 34%-os elméleti hozammal számolhatunk.
8a hozamának további növelése érdekében az alkalmazott dietil-amin nukleofil reagens arányát csökkentettem. Az 1,1'dijód-ferrocénhez képest tízszeres mennyiségû nukleofil reagens helyett csupán háromszoros felesleget alkalmazva sikerült növelnem 8a mennyiségét. A maximális hozamot 32 óra elteltével értem el (3.táblázat 12.sor), ami a gázkromatográfiás adatok alapján számolva elméletileg 42%-nak adódott. Az oszlopkromatográfiás elválasztás után 35%-os kitermeléssel jutottam a termékhez.
Ezen tapasztalatokra alapozva, egyéb szekunder aminok alkalmazásával igyekeztem további 1'-jód-ferrocénkarboxamidokat, vagy -glioxamidokat elõállítani, melyeket egy következõ homogénkatalitikus átalakítás során kiindulási anyagként felhasználhatok. A dietil-amin esetén alkalmazott stratégia a legtöbb esetben hatékonynak bizonyult, azonban a szelektivitás területén jelentõs eltérések is adódtak (4. táblázat). A dibutil-amin használata során kapott eredmény nem okozott különösebb meglepetést, ugyanis az alacsony hõmérséklet és a csökkentett nukleofil reagens arány mellett megvalósított reakció nagyon hasonló hozamokhoz vezetett (4. táblázat 3.sor), mint azt a dietil-amin esetében tapasztaltam. Morfolin és piperidin alkalmazásakor azonban már jelentõs eltérések mutatkoztak. A 60°C-on kivitelezett reakcióban, az eddigiektõl eltérõen már az 1'-jód-ferrocénglioxamid (9c, 9d) a fõtermék, de mellette jelentékeny arányban jelen van az 1'-jód-ferrocénkarboxamid (8c, 8d) is (4. táblázat 5. és 7. sor). Ez utóbbi két esetben közepes hozammal sikerült elkülönítenem az említett kettõs karbonilezett származékokat.
Alacsonyabb hõmérsékleten, illetve a nukleofil reakciópartnerként alkalmazott szekunder amin feleslegének csökkentésével sikerült a következõ 1'-jód-ferrocénkarboxamidokat, illetve -glioxamidokat közepes hozammal elõállítanom: 1'-jód-N,N-dietil-ferrocénkarboxamid (8a), 1'-jód-N,N-dibutil-ferrocénkarboxamid (8b), 1'-jód-morfolino-ferrocénglioxamid (9c), 1'-jód-piperidino-ferrocénglioxamid (9d). A továbbiakban ezek további homogénkatalitikus reakcióit vizsgáltam.
(A 9b és 11a termékeket nem különítettem el, ezeket a vegyületeket a reakcióelegyek GC-MS vizsgálatával azonosítottam. 11b-d képzõdését nem tapasztaltam.)
A fent említett 1'-jód-ferrocénszármazékok elõállításának célja volt, hogy újabb homogénkatalitikus reakciók segítségével heterodiszubsztituált ferrocénszármazékokat szintetizáljak. Az elõzõ fejezetben ismertetett karbonilezési reakciókban legjobb hozammal nyert 1'-jód-származékokat: 8a-t, valamint 9c-t és 9d-t különbözõ palládium-katalizált reakciók: aminokarbonilezés, NaBPh4 jelenlétében lejátszódó karbonilatív kapcsolás és Stille-kapcsolás segítségével alakítottam tovább. A két homogénkatalitikus lépés viszonylag egyszerû lehetõséget biztosít a két különbözõ szubsztituenssel helyettesített ferrocénszármazékok, pl. 1,1'-diamidok elõállítására szemben az eddig alkalmazott, bonyolultabb módszerekkel (ld. 1.3. fejezet).
Elsõként a 8a amidot reagáltattam karbonilezési
reakcióban különbözõ szekunder aminokkal (2.5 egyenlet). 40 bar CO nyomáson
100°C-on 8 óra alatt teljes átalakulással megkaptam a megfelelõ "vegyes" diamidokat
( 10ab, 10ac, 10ad) (izolált hozam:
92%, 86%, 77%). Kettõs karbonilezett termék keletkezését még alacsonyabb
hõmérsékleten (60-

(2.5)
A 8a, 9c és 9d származékok Stille-kapcsolásával vinil-ferrocén származékokat (13a, 14c, 14d) állítottam elõ vinil-tributil-sztannán reakciópartner segítségével Pd(PPh3)4 jelenlétében (2.6 egyenlet). A reakciókörülményeket a Szerves Kémia Tanszéken, szteránvázas alkenil-jodidok átalakítása során nyert tapasztalatok alapján [78] választottam meg. A reakció könnyedén lejátszódott, az átalakulás már 3 óra elteltével minden esetben teljes volt.
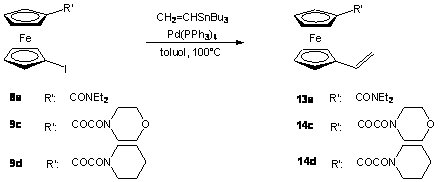
Végül egy - szintén a Pannon Egyetem Szerves Kémia Tanszékén kidolgozott fenil-ketonokat eredményezõ - karbonilatív kapcsolás [71] révén állítottam elõ a 15a származékot (2.7 egyenlet). 8a-t reagáltattam ekvimoláris mennyiségû NaBPh4-al szén-monoxid atmoszférában Pd(PPh3)4 jelenlétében. A reakció elõrehaladását gázkromatográfiás úton követtem. 6 óra reakcióidõt követõen az átalakulás számottevõen már nem változott, ekkor az átalakulás 82% volt. A terméket 78%-os hozammal sikerült elkülönítenem.
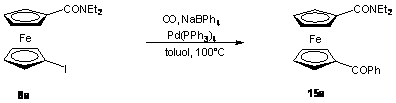
(2.7)
Az eredmények alapján elmondható, hogy a kidolgozott módszer jól alkalmazható változatos szerkezetû ferrocénvegyületek elõállítására. Bár az 1'-jód-ferrocénkarboxamid és 1'-jód-ferrocénglioxamid származékok csak közepes hozammal izolálhatók az 1,1'-dijód-ferrocén aminokarbonilezésének termékeibõl, a második homogénkatalitikus lépés rendkívül szelektíven és jó hozammal szolgáltatja a kívánt termékeket.
Az elõzõ fejezetben bemutattam, hogyan állítottam elõ különbözõ heterodiszubsztituált ferrocénszármazékokat, köztük a 10ab, 10ac, 10ad diamidokat két lépésben.
Felmerült a kérdés, ilyen típusú vegyületeket nem lehet-e 1,1'-dijód-ferrocénbõl egy szintetikus lépésben, két különbözõ nukleofil reagens együttes jelenlétében szintetizálni. A reakciót a kettõs karbonilezés elkerülése érdekében 100°C-on kiviteleztem. Egyébként a jód-ferrocén aminokarbonilezésénél optimálisnak talált körülmények között dolgoztam. Ekkor a korábbi tapasztalatok alapján csupán a 2.8 egyenletben feltüntetett három termék - két szimmetrikusan (10x és 10y) és egy nem szimmetrikusan szubsztituált diamid (10xy) - képzõdésével kellett számolnom.

A karbonilezés elõrehaladását gázkromatográfiás úton követtem. Az eredmények azt mutatták, hogy a reakció kimenetele erõsen függ a nukleofil reagensként használt aminok természetétõl (5. táblázat).
Amikor a kétféle nukleofil reagenst azonos mennyiségben alkalmaztam, a hasonló sztérikus és elektronikus tulajdonságokkal rendelkezõ aminok esetén, amilyen a dietil-amin - dibutil-amin pár (5. táblázat 1. sor), a termékek a statisztikailag várható 10ab / 10a / 10b = 2 / 1 / 1 arányban keletkeztek, ilyen esetekben a vegyes amidok (10xy) aránya nem növelhetõ. A többi vizsgált amin pár hasonló körülmények között lejátszódó reakciójában általában a nem szimmetrikusan szubsztituált vegyület (10xy) mellett csak az egyik szimmetrikus diamid képzõdött (5. táblázat 2., 7., 8., 10. és 12. sor). Kivételt képez a piperidin jelenlétében lejátszódó két reakció, ahol a piperidin olyan sikeresen verseng a dietil-aminnal (5. táblázat 4.sor) vagy a dibutil-aminnal (15. sor), hogy mindkét reakcióelegyben kizárólag 10c jelenlétét figyeltem meg. Külön ki kell emelnem a dietil-amin és morfolin valamint a dibutil-amin és morfolin jelenlétében lejátszódó aminokarbonilezést (5. táblázat 7. és 10. sor), ahol a két amin 1/1 arányú elegyének
5. táblázat 1,1'-dijód-ferrocén karbonilezése két különbözõ amin együttes jelenlétében
|
Sorsz. |
R1R2NH (x) |
R3R4NH (y) |
R1R2NH / |
Reakcióidõ |
Konv.(%) |
szelektivitás(%) (izolált hozam) (%) |
||
|
R3R4NH |
(h) |
10xy |
10x |
10y |
||||
|
|
HNEt2 (a) |
HNBu2 (b) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
morfolin (c) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
piperidin (d) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
piperidin (d) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
piperidin (d) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
H2NBu (e) |
|
|
|
|
|
|
|
|
HNEt2 (a) |
H2NBu (e) |
|
|
|
|
|
|
|
|
HNBu2 (b) |
H2NBu (e) |
|
|
|
|
|
|
|
|
HNBu2 (b) |
H2NBu (e) |
|
|
|
|
|
|
|
|
HNBu2 (b) |
H2NBu (e) |
|
|
|
|
|
|
|
|
morfolin (c) |
piperidin (d) |
|
|
|
|
|
|
|
|
morfolin (c) |
piperidin (d) |
|
|
|
|
|
|
|
|
morfolin (c) |
H2NBu (e) |
|
|
|
|
|
|
|
|
morfolin (c) |
H2NBu (e) |
|
|
|
|
|
|
|
|
morfolin (c) |
H2NBu (e) |
|
|
|
|
|
|
aGC alapján; Reakciókörülmény: 1,1'-dijód-ferrocén (7) (1 mmol), R1R2NH + R3R4NH (10mmol), Et3N (1 mL), Pd(OAc)2 (0,1 mmol), PPh3 (0,2 mmol), toluolban
alkalmazásakor is 90, illetve 88%-os szelektivitással képzõdött a heterodiszubsztituált vegyület (10ac és 10bc). A többi esetben a dietil-amin dibutil-amin reagens pár kivételével a kevésbé reakcióképes amin arányának növelésével sikerült a nem szimmetrikusan szubsztituált származékok képzõdését növelni (5. táblázat 2.-15. sor).
Úgy tûnik azonban, hogy nem kizárólag az aminok reakciókészsége, vagy bázicitása befolyásolja az aminokarbonilezés szelektivitását. Jól szemlélteti ezt a következõ kísérlet. Jód-ferrocént reagáltattam ekvimoláris mennyiségû, jód-ferrocénre nézve egyenként ötszörös feleslegben lévõ morfolin és dietil-amin elegyével aminokarbonilezési körülmények között 100°C-on (2.8).

A két termék: az N,N-dietil-ferrocénkarboxamid (16a) és a morfolino-ferrocénkarboxamid (16c) 1/7 arányban keletkezett. Ez alapján úgy tûnik, hogy annak ellenére, hogy a morfolin bázicitása kisebb, mint a dietil-aminé, mégis nagyobb a reakciókészsége. Ha azonban ugyanezen amin pár szintén 1/1 arányú elegyét 1,1'-dijód-ferrocénnel reagáltatjuk, azt tapasztaljuk, hogy 10c nem keletkezik (5. táblázat 2. sor) annak ellenére, hogy az elõzõ reakció alapján a morfolin tûnt reakcióképesebbnek. Valamilyen oknál fogva a morfolinnal képzõdõ szimmetrikus diamid keletkezése kedvezõtlen. Némileg egybevág ezzel a megfigyeléssel a 2.3. fejezetben leírt tapasztalat, melyben azt láthatjuk (4. táblázat), hogy az 1,1'-dijód-ferrocén szekunder aminok jelenlétében lejátszódó aminokarbonilezési reakcióiban 100°C-on kizárólag a megfelelõ ferrocén-diamid keletkezik, kivéve a morfolin esetében, ahol ugyanannyi idõ elteltével 1'-jód-ferrocén-karboxamid jelenléte is megfigyelhetõ volt (4. táblázat 4. sor).
Más aminok morfolinnal egyidejû alkalmazásakor is hasonló eredményt tapasztaltam, mint dietil-amin esetén, ami szintén azt mutatja, hogy a morfolinnal képzõdõ szimmetrikus diamid keletkezése némileg gátolt (5. táblázat 11.-14. sor. ).
Nagyon sok reakcióban tehát a két amin arányának megfelelõ megválasztásával elérhetõ, hogy a nem szimmetrikus 1,n'-ferrocéndikarboxamidok aránya messze a legmagasabb legyen. Így ez az eljárás egy meglehetõsen egyszerû és hatékony alternatívát kínál e vegyületek szintézisére (vö. 1.10 egyenlet).
Mint azt a 1.2. fejezetben láthattuk, a ferrocén amidokat elõszeretettel alkalmazzák különbözõ kémiai szenzorokként, molekula- és ionreceptorokként. Ezen alkalmazásoknál nagyon sokszor kulcsszerepet játszik a receptor (ferrocén származék) és a "vendég"molekula, vagy ion között kialakuló hidrogénkötés. Megfelelõ szubsztituensek esetén magán a ferrocén származékon belül is kialakulhatnak hidrogénkötések, melyek döntõen befolyásolják a molekula szerkezetét, viselkedését. Ezen hidrogénhidak jelentõségét tekintve nem meglepõ, hogy számos kutatócsoport írta le megfigyeléseit ezzel kapcsolatban. [24, 79-81]
Az
elõzõekben bemutatott módszerek segítségével elõállított diamidok (2.2 ábra) 1H-NMR
spektrumainak vizsgálatakor magam is számos érdekes megfigyelésre jutottam. Az egy
N-szubsztituált és egy N,N-diszubsztituált amidocsoportot
tartalmazó (10ae, 10be, 10ce), illetve a két N,N-diszubsztituált
amidocsoportot tartalmazó diamidok (10ac,
10ab, 10ad, 10cd, 10a, 10b, 10c, 10d) spektrumának összehasonlításában az
egyik szembetûnõ és nyilvánvaló eltérés az elõbbiekben az NH proton jelenléte,
amely 8 ppm fölötti tartományban található (6. táblázat 1-3 sor). Az amid NH
protonjának ilyen magas kémiai eltolódása 10ae,
10be, 10ce esetében hidrogénkötés létrejöttére utalhat. A másik
figyelemre méltó eltérés a két N,N-diszubsztituált
amidocsoportot tartalmazó "vegyes" diamidok (10ab, 10ac, 10ad, 10cd) spektrumával összehasonlítva a ferrocén váz Cp potonjaiban
mutatkozott. Ezen diamidok ciklopentadienil gyûrûinek 2,5; 3,4;
![]()
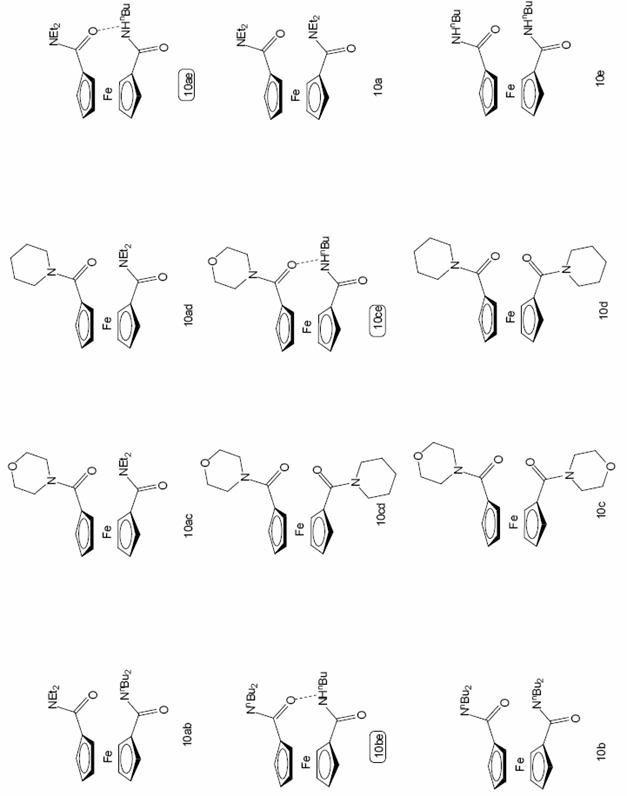
protonjaival. Ebbõl az látszik,hogy az egyik ciklopentadienil gyûrûn lévõ szubsztituens nincs befolyással a másik Cp gyûrû protonjaira. Merõben más a helyzet azonban az egy N-szubsztituált és egy N,N-diszubsztituált amidocsoportot tartalmazó diamidok esetében. A 10ae, 10be, 10de vegyületek 1H-NMR spektrumát vizsgálva az elõbb tárgyalt tartományban, a jelek összeolvadását figyelhetjük meg. Elvégezve az iménti összehasonlítást pl. 10ae és a megfelelõ szimmetrikus diamidok (10a és 10e) között, a következõ eltolódásokat láthatjuk: 10ae spektrumában: 4,60(s, 2H); 4,36(brs, 4H) és 4,32(s, 2H), 10a spektrumában: 4,63(s, 4H); 4,36(s, 4H) és 10e spektrumában: 4,46(s, 4H); 4,30(s, 4H) a ciklopentadienil gyûrûk protonjainak eltolódása. Az adatokból jól látható az eltérés, amely a késõbbi vizsgálatok alapján úgy tûnik, hogy szintén az intramolekuláris hidrogénhíd létrejöttének köszönhetõ.
6. táblázat 1,1'-Ferrocén-bisz-karboxamidok jellemzõ spektroszkópai adatai
|
Sorsz. |
|
nNHa (cm-1) |
Amid-Ia (cm-1) |
Amid-IIa (cm-1) |
dNHc (ppm) |
dCpc (ppm) |
|
|
10ae |
|
1613 |
|
|
|
|
|
10be |
|
1610 |
|
|
|
|
|
10ce |
|
|
|
|
|
|
|
10ab |
|
|
|
|
|
|
|
10ac |
|
|
|
|
|
|
|
10ad |
|
|
|
|
|
|
|
10cd |
|
|
|
|
|
|
|
10a |
|
|
|
|
|
|
|
10b |
|
|
|
|
|
|
|
10c |
|
|
|
|
|
|
|
10d |
|
|
|
|
|
|
|
10e |
|
|
|
|
|
aIR (KBr)
b 1H-NMR (400MHz,CDCl3)
Mindezen figyelemre méltó megfigyelések arra sarkalltak, hogy a 10ce vegyületet részletes spektroszkópiai vizsgálatoknak vessem alá mind szilárd, mind pedig oldott állapotban.
Munkám során a keletkezett termékbõl több ízben próbáltam egykristályt növeszteni. Egyedül 10ce-t sikerült diklór-metán-dietil-éter oldószerelegybõl szerkezetvizsgálatra alkalmas egykristály formájában kinyernem. A molekula szerkezetét a Debreceni Egyetem Kémiai Tanszékcsoportjának röntgenkrisztallográfiai laboratóriumában Dr. Bényei Attila határozta meg. A kapott kötéstávolság és kötésszög értékekbõl a kristályszerkezet az ORTEP és WINGX-97 programcsomag segítségével készült el (2.3 ábra).
A H42.O1 2,18(5) Ǻ és az N42.O1 2,933(6) Ǻ atomok közötti kötéstávolság, illetve az N42-H42.O1 174(4)° kötésszög jól jellemzi a molekulában kialakult erõs intramolekuláris hidrogénkötést. A ferrocén molekularészlet központi vasatomja és a hozzá koordinálódó ciklopentadienil szenek Fe-C távolsága 2,010-2,050 Ǻ, ez rövidebb az adatbázisokban egyéb ferrocénszármazékoknál található 2,080 Ǻ értéknél [82, 83]. A molekula szerkezetét tovább stabilizálja az O1 illetve O41 atomok és a morfolin valamint a Cp gyûrû térben közel kerülõ hidrogénatomjai között kialakuló gyenge hidrogénkötés.
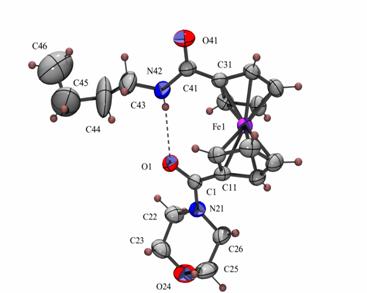
2.3. ábra 10ce vegyület röntgendiffrakciós szerkezetének ORTEP rajza
kötéstávolság (Ǻ), kötésszög(°): Fe-C 2,013-2,052; C1-O1 1,237(5); C1-C11 1,494(5); C1-N21 1,339(5); C31-C41 1,469(5); C41-O41 1,235(5); C41-N42 1,318(6); N42-C43 1,463(7); O1-N42 2,938(5); N42-H42-O1 174(6)
A 10ce KBr pasztilla segítségével felvett infravörös spektrumán 3252 cm-1-nél található a n(NH) vegyértékrezgési sáv. 1642 cm-1-nél találjuk a hidrogénhídban nem szereplõ C=O és 1593 cm-1-nél az amid NH-val hidrogénkötésben lévõ C=O vegyértékrezgési sávjait, az úgynevezett amid-I sávokat egyenlõ intenzitással. Az amid-I sáv nagyobb részben a C=O vegyértékrezgésbõl, kisebb részben ugyan, de a C-N vegyértékrezgésbõl is ered és 1800-1630 cm-1 intervallumban jelenik meg. Pontos helye az amidocsoport szubsztituenseitõl függ, de tudjuk, hogy a karbonilcsoport oxigénjével kialakuló hidrogénhidak csökkentik az amid-I sáv helyét. 1552 cm-1-nél jelenik meg az amid-II sáv, melyben a síkban deformációs NH rezgésmód dominál, de C-N vegyértékrezgésmód is jelentõs szerepet játszik.
A szilárd fázisú vizsgálatok megbízhatóan igazolták a molekula két szubsztituense között kialakult hidrogénkötést. A molekula oldott állapotban lévõ szerkezetének tisztázását infravörös és 1H-NMR spektroszkópiai módszerek segítségével végeztem el.
Irodalmi adatok szerint hidrogénkötésben részt nem vevõ amid NH csoportok nNH vegyértékrezgési sávja 3400 cm-1 fölötti hullámszám tartományban jelentkezik a molekula infravörös spektrumában, kémiai eltolódása pedig 5,5 és 7,5 ppm között található az 1H-NMR spektrumon. Ugyanakkor ha az amid NH hidrogénkötésben van, a nNH vegyértékrezgési sáv 3400 cm-1 alatt, míg protonjának jele 7,5 ppm fölötti kémiai eltolódásnál keresendõ. Mindezek mellett az NH protonok kémiai eltolódásának kis mértékû (-2 - -4 ppb K-1) hõmérsékletfüggése (ΔδH) jelentheti egyrészt hidrogénhíd hiányát, illetve annak erõs mivoltát. Ezen irodalmi megfigyeléseket [80] alapul véve végeztem el 10ce oldat fázisú szerkezetvizsgálatát.
10ce diklórmetán oldatban felvett IR spektrumán két különbözõ nNH vegyértékrezgési sáv található, egy gyengébb 3488 cm-1-nél és egy erõsebb sáv 3272 cm-1-nél. Ennek magyarázata a kétféle - hidrogénkötésben lévõ és szabad - amid NH egyidejû jelenléte oldott állapotban. Értelemszerûen a nagyobb hullámszámnál jelentkezõ jel tartozik a hidrogénhíd mentes formához (2.9 egyenlet B szerkezet), míg az alacsonyabb hullámszámú sáv a hidrogénhidas forma (2.9 egyenlet A szerkezet) létezését támasztja alá. Ez utóbbi sáv nagyobb intenzitása jelzi az A szerkezet dominanciáját az adott körülmények között.
A hidrogénkötés jelenléte jól megfigyelhetõ a karbonilcsoport jellemzõ vegyértékrezgési sávjainak megfelelõ hullámszámtartományban (amid-I sáv) is. A karbonilcsoport oxigénjével kialakult hidrogénhidak jelentõsen csökkentik a C=O vegyértékrezgési sáv hullámszámát. Ennek megfelelõen két n(C=O) vegyértékrezgési, vagy amid-I sávot látunk az oldat IR spektrumán. Egy intenzívebb, szélesebb sávot 1646 cm-1-nél, amely megfelel az A forma hidrogénhídban részt nem vevõ CO, illetve a B forma mindkét karbonilcsoportjának. Körülbelül 30 cm-1-rel alacsonyobb hullámszámnál, 1614 cm-1-nél találjuk az A forma hidrogénhidas karbonilcsoportjának jelét. Az A és B forma együttes jelenlétének megfelelõen az amid-II sávokból is kettõt találunk, egyenként 1548 és 1523 cm-1-nél.
10ce NH protonjának kémiai eltolódása 30°C-on deutero-kloroformban 8,32 ppm, toluol-d8-ban 8,47 ppm. A toluolos oldat 15-szörös hígítása az NH proton kémiai eltolódásának csupán 0,02 ppm-es csökkenését idézte elõ, ami valószínûsíti a feltételezett intramolekuláris kölcsönhatás létrejöttét a molekula két szubsztituense között. Az NH proton helyének hõmérsékletfüggését vizsgálva toluolban, 303-353 K között, a ΔδH = -14,3 ppb K-1-nek adódott, amely egy közepesen erõs dinamikus hidrogénkötésnek felel meg.
Mivel az irodalom a ferrocénkarbonsav-amidokat és -diamidokat kiváló anion receptorokként tartja számon, kézenfekvõ volt, hogy 10ce infravörös és 1H-NMR vizsgálatait kloridion jelenlétében is elvégezzem. Az anionok hidrogén akceptor sajátságát figyelembe véve, kloridionok jelenléte várhatóan erõs befolyást gyakorol az intramolekuláris hidrogénhíd kialakulására, illetve megszûnésére (2.9. egyenlet).
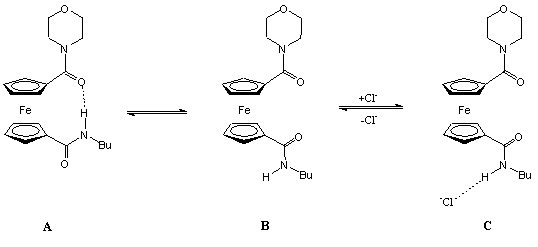 (2.9)
(2.9)
n(NH) 3272 cm-1 3448 cm-1 3285 cm-1
amid-I 1646, 1614 cm-1 1646 cm-1 1654, 1642cm-1
amid-II 1548 cm-1 1523 cm-1 1548 cm-1
A tetra-n-butil-ammónium-klorid formájában hozzáadott kloridion valóban markáns változásokat idézett elõ mind az infravörös, mind pedig az 1H-NMR spektrumokban (7. táblázat). A szabad amid NH 3448 cm-1-es vegyértékrezgési sávjának eltûnése mellett az amid-I sávok hasadását tapasztaltam. Négyszeres kloridion felesleget elérve ez utóbbi jelek szélesedése, egybecsúszása miatt alkalmatlanná váltak a molekulaszerkezet változásainak precíz követésére. Az 1550 cm-1 körül jelentkezõ hidrogénkötésben résztvevõ amid amid-II sávjának intenzitásnövekedése mellett a szabad forma (B) amid-II sáv-intenzitásának folyamatos csökkenését figyeltem meg, ami szintén a B → C átalakulás lejátszódására utal.
A kloridion-koncentráció növelésének hatására a 1H-NMR felvételeken is ugyanaz a tendencia figyelhetõ meg, mint az infravörös spektrumokon. Az NH proton kémiai eltolódásának növekedése jelzi, hogy egyre több NH proton vesz részt hidrogénhíd kialakításában (B → C forma). Ezen túlmenõen magas kloridion koncentrációnál a Cp protonok az addig megfigyelt három szingulett helyett négy jellé különülnek. Ez a jelenség valószínûleg az intramolekuláris hidrogénhíd helyett kialakuló intermolekuláris hidrogénhíd megjelenésének köszönhetõ (C forma). A C szerkezetben a két gyûrû szubsztituensei ismét "függetlenek" egymástól, ugyanúgy, mint a kizárólag N,N-diszubsztituált amid csoportot tartalmazó diszubsztituált ferrocénszármazékok esetén, ahol nincs lehetõség intramolekuláris hidrogénhíd képzõdésére.
Említést érdemel, hogy a két N-szubsztituált amidocsoportot tartalmazó ferrocénszármazék (10e) spektrumaiban oldott állapotban hidrogénhíd kialakulására utaló jelet - csakúgy mint mások [24] - nem tapasztaltam.
7. táblázat 10ce jellemzõ infravörös és 1H-NMR spektroszkópiai adatai kloridion jelenlétében
|
Cl- / 10ce |
nNHa (cm-1) |
Amid-Ia (cm-1) |
Amid-Iia (cm-1) |
dNHc (ppm) |
dCpc (ppm) |
|
|
3252b |
1640, 1593b |
1552b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a c(10ce)=3x10-2M, diklór -metánban 25°C-on b Szilárd fázisú, KBr-ben c c(10ce)=5x10-2M CDCl3-ban 30°C-on
A ferrocénszérmazékokat stabilitásuk, kedvezõ elektrokémiai tulajdonságai miatt elõszeretettel alkalmazzák elektrokémiai bioszenzorok építõkövének (1.2.2. fejezet). Ennek következtében számos módszert dolgoztak ki különbözõ biomolekulák ferrocénvázhoz való csatolására [84]. Ezen molekulák közül is kitüntetett érdeklõdés övezi az 1,n'-diszubsztituált ferrocenoil-aminosavakat és peptideket, melyek még oldott állapotban is viszonylag rendezett molekulaszerkezettel rendelkeznek [85, 86]. Az ilyen diszubsztituált ferrocénszármazékok két ciklopentadienil gyûrûjéhez túlnyomó többségben két azonos aminosav, vagy peptid kapcsolódik. A legritkább esetben találkozunk olyan heterodiszubsztituált ferrocenoil-aminosav, vagy peptid származékokkal, melyekben a ferrocén ciklopentadienil gyûrûi két különbözõ aminosav, vagy peptid származékkal helyettesítettek. Ennek oka valószínûleg az eddig alkalmazott szintézisek összetettségében, bonyolultságában keresendõ. Mindössze három példát találtam az irodalomban ilyen heterodiszubsztituált származékok elõállítására, melyeket az 1.4 fejezetben tárgyaltam.
Tekintetbe véve az említett vegyületcsalád mind szimmetrikusan helyettesített, mind heterodiszubsztituált képviselõinek jelentõségét, lényeges lehet egy egyszerû, a korábbiaknál hatékonyabb elõállítási mód kidolgozása. Az általam alkalmazott homogénkatalitikus palládium-katalizált aminokarbonilezés ennek megoldására kínál egy elegáns lehetõséget.
A
korábbi kutatási eredményeimre támaszkodva a 2.10 egyenletnek megfelelõen
1,1'-dijód-ferrocént (7)
aminokarbonilezési reakcióba vittem két különbözõ aminosav-észtert ekvimoláris
mennyiségben tartalmazó eleggyel. Katalizátorként a jól bevált Pd(OAc)2
és két ekvivalens trifenil-foszfán elegyét használtam. A karbonilezésnél 40 bar
szén-monoxid nyomást és
(Meg kell jegyeznem, hogy az aminokarbonilezés minden valószínûség szerint alkalmas a szimmetrikus 17a-d származékok szelektív szintézisére egyetlen aminosav-észter reakciópartner alkalmazása esetén. Az én célom a heterodiszubsztituált vegyületek elõállítása volt, tehát az egyetlen nukleofil reagens jelenlétében lejátszódó karbonilezést nem vizsgáltam. A 17a-d vegyületek mint melléktermékek képzõdtek, ezért ezekre a vegyületekre hozamokat nem adtam meg a 4. fejezetben.)
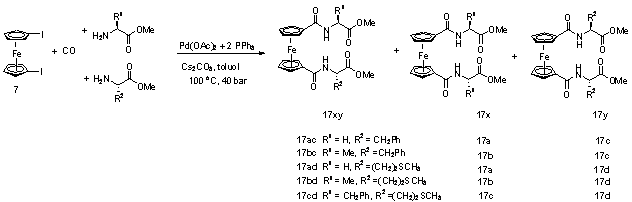
(2.10)
Az esetek többségében a heterodiszubsztituált ferrocénkarbonsav származékok és a szimmetrikusan helyettesített társaik körülbelül a statisztikailag várt 17xy:17x:17y = 2:1:1 arányban keletkeztek (6. táblázat 2-4 sor). Ettõl az eredménytõl némileg eltér a glicin-metil-észter-fenilalanin-metil-észter nukleofil reagenspár használatakor kapott termékarány. A glicin-metil-észterbõl képzõdött szimmetrikus ferrocenoil aminosavészter származék Fe[C5H4-CO-Gly-OMe]2 (17a) mennyisége valamiért alul marad fenilalanin-metil-észterrel képzõdött párjáétól Fe[C5H4-CO-Phe-OMe]2 (17c), ugyanakkor a kívánt heterodiszubsztituált származék [MeO-Phe-CO-C5H4-]Fe[C5H4-CO-Gly-OMe] (17ac) részaránya itt a legnagyobb (8. táblázat 1.sor).
8. táblázat Az 1,1'-dijód-ferrocén (7) és két különbözõ aminosav-észter reakciójában kapott termékek (17) hozama
|
Sorsz. |
Termék Hozam (%) |
||
|
|
17xy |
17x |
17y |
|
|
17ac 47 |
17a 8 |
13c 24 |
|
|
17bc 27 |
17b 13 |
17c 13 |
|
|
17ad 39 |
17a 24 |
17d 19 |
|
|
17bd 40 |
17b 18 |
17d 16 |
|
|
17cd 52 |
17c, |
17d 19 |
Reakciókörülmények: 1,1'-dijód-ferrocén (7) (0,5 mmol), Pd(OAc)2 (0,05 mmol), PPh3
(0,1 mmol), két különbözõ aminosavészter-hidroklorid (2 x 0,6 mmol), Cs2CO3
(2,2 mmol), toluol (10 mL),
Bár a reakcióban három, egymáshoz nagyon hasonló szerkezetû termék képzõdik, a komponensek oszlopkromatográfiás módszerrel szétválaszthatók (szilikagél, toluol-metanol, 7:1). A 8. táblázat izolált hozam adatai alapján elmondható, hogy a szóban forgó eljárás heterodiszubsztituált 1,1'-ferrocenoil-bisz-aminosav-észterek elõállításának egy meglehetõsen egyszerû módja.
Korábbi közlemények ilyen típusú ferrocéndikarboxamid származékok rendezett szerkezetérõl számolnak be, melyet a két szemközt lévõ észter C=O és amid NH csoportok között kialakult 1-1 intramolekuláris hidrogénhíd stabilizál (2.4 ábra) [24, 80, 81, 87].
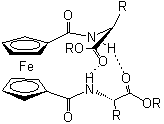
2.4 ábra
Az általam elõállított új vegyületek 17ac, 17cd, 17ad és 17bd esetében az infravörös és 1H-NMR spektroszkópiai adatok: az NH protonok viszonylag magas, 7,5 ppm fölötti eltolódása, illetve a 3400 cm-1 alatt jelentkezõ nNH vegyértékrezgési sáv, szintén az elõbb vázolt szerkezet kialakulását támasztja alá.
A módszer továbbfejlesztésével lehetõség nyílhat tetszõleges, akár két különbözõ peptidlánc kiépítésére a ferrocén két ciklopentadienil gyûrûjén. Amennyiben két eltérõ védõcsoporttal rendelkezõ aminosav-észtert alkalmazunk az aminokarbonilezési reakció nukleofil reakciópartnereként, úgy a kapott vegyes ferrocéndikarboxamid származékok eltérõ módon védett aminosav láncai különbözõ módszerekkel egyenként hidrolizálhatóak, illetve aktiválhatóak.
Az eddigiekben a heterodiszubsztituált 1,1'-ferrocén származékok elõállításának két homogénkatalitikus lehetõségét mutattam be. A ferrocén Cp gyûrûinek nagy reakcióképessége miatt lehetõség van arra is, hogy egyszerû, klasszikus szerves kémiai módszerek bevonásával könnyedén heterodiszubsztituált ferrocénszármazékokhoz jussunk. Így állítottam elõ az Eötvös Lóránd Tudományegyetem Peptidkémiai Kutatócsoportjával és Szervetlen Kémiai Tanszékével való együttmûködés keretein belül N-ferrocenoil-glicin-metil-észter (2a) Friedel-Craft acilezésével 1'-acetil-N-ferrocenoil-glicin-metil-észtert (18) (2.11 egyenlet).
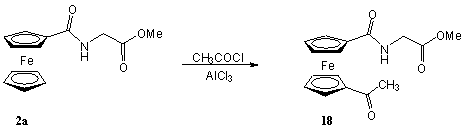
(2.11)
A szintézis második lépését Eisenthal módszere szerint hajtottam végre [88], melynek eredményeként a teljes átalakulásnak köszönhetõen egy egyszerû feldolgozást követõen, tisztán kaptam meg a kívánt acilezett ferrocénszármazékot (18) (hozam: 99%).
A karbonilezési reakciók kiindulási anyagaként szolgáló jód-, illetve dijód-ferrocén elõállításához felhasznált ferrocén a Flukától, a jód a Reanaltól származott, míg a n-butil- és t-butil-lítium oldatok Aldrich márkájú anyagok voltak.
A homogénkatalitikus
karbonilezésben használt katalizátor prekurzor palládium-acetát és trifenil-foszfán
a Fluka által forgalmazott vegyszer volt. Az egyéb kapcsolási reakciók
katalizátoraként szolgáló Pd(PPh3)
A reakciókban bázisként alkalmazott trietil-amin Carlo Erba, a cézium-karbonát Aldrich termék volt. A trietil-amint felhasználás elõtt argon alatt átdesztilláltam és inert körülmények között tároltam. A nukleofil reagensek közül az aminok Fluka vegyszerek voltak, az aminosavésztereket szintén Fluka gyártmányú aminosavakból, tionil-kloriddal (Reanal) és metanollal készítettem.
A Stille-kapcsolás vinil-tributil-sztannán reagense Aldrich, a NaBPh4-os kapcsolás reagense Fluka, az N-ferrocenoil-glicin-metil-észter acilezési reakciójában használt alumínium-klorid Fluka, az acetil-klorid Reanal reagens volt.
A ferrocénvázas termékek szétválasztására szolgáló oszlopkromatográfiás eljárás során állófázisként alkalmazott alumínium-oxid (BrockmannII, neutrális) Spektrum 3D Kft. által forgalmazott anyag.
A reakcióelegyek feldolgozása során használt sósav, nátrium-hidrogén-karbonát és nátrium klorid oldatokat szintén Spektrum 3D-s vegyszerekbõl készítettem.
A kísérlethez használt oldószerek Fluka, Reanal és Spektrum 3D analitikai tisztaságú vegyszerek voltak.
A legtöbb reakció oldószereként használt toluolt az analitikai tisztaságú oldószer argon alatti desztillációjával készítettem, benzofenon és fém nátrium segítségével.
A kísérleti munka folyamán a vegyszerek, oldószerek beméréséhez és a reakciótérhez, mint inert rendszerhez szükséges feltételek biztosítására szolgáló argon gázt a Messer Hungarogáz Kft. forgalmazta. Közvetlenül a használat elõtt az argonból "DEDUX" töltetû oszlopon a nyomokban lévõ oxigént eltávolítottam, valamint a gázt szilikagéllel és kálium-hidroxiddal töltött csöveken keresztülvezetve szárítottam.
A karbonilezési reakcióhoz szükséges szén-monoxid szintén a Messer-Hungarogáz Kft. terméke volt.
Gázbevezetõvel,
szeptumos feltéttel és csepegtetõtölcsérrel ellátott reaktorba bemértem 15 mmol
(
Az
1 órás keverés közben egy Schlenk edénybe bemértem
Az 1 órás 0°C-os keverést követõen a reakcióelegyet cseppfolyós nitrogén - aceton eleggyel -78°C-ra hûtöttem és ezen a hõmérsékleten cseppenként hozzáadtam a jódoldatot kb. 40 perc alatt. Ezt követõen a reakcióelegyet további 1 órán át kevertem, majd a hûtõfürdõ eltávolítása után hagytam a reakcióelegyet szobahõfokra melegedni.
A reakció végén az elegyhez 100 ml vizet adtam a feleslegben maradt t-BuLi reagens elbontása érdekében, majd választótölcsér segítségével elkülönítettem a szerves és a vizes fázist. Ezután a vizes fázist 30 ml dietil-éterrel 3 ízben extraháltam. A szerves fázisokat egyesítettem és mostam vízzel, telített Na2S2O3 és telített NaCl oldattal. Ezután a szerves fázist Na2SO4-al szárítottam, majd az oldószert vákuumdesztillációval eltávolítottam.
A tiszta jód-ferrocént a maradék ferrocén szennyezéstõl oszlopkromatográfiával választottam el. Állófázisként Brockmann-II, neutrális alumínium-oxidot használtam, a kromatográfia eluense n-hexán volt.
Egy gázbevezetõvel, szeptumos feltéttel és csepegtetõtölcsérrel ellátott reaktort argon alá helyeztem majd a szeptumos feltéten keresztül bemértem 24,6 mmol (3,7 ml) tetrametil-etilén diamint (TMEDA). Ehhez hozzáadtam a szeptumon keresztül 3,4 ml n-hexánt. Ezután a csepegtetõtölcsér segítségével szobahõmérsékleten, keverés közben cseppenként hozzáadtam 24,6 mmol n- BuLi-ot 1,5 M-os pentános oldat formájában (15,3 ml). A beadagolás befejeztével az elegyet további 10 percig szobahõfokon kevertem.
Ezalatt
egy Schlenk edénybe bemértem 10 mmol (
A 10 perces keverés elteltével az elõzekben elkészített ferrocén hexános oldatát a csepegtetõ tölcsér segítségével hozzáadtam a n-BuLi-TMEDA komplex oldatához. A dilítium vegyület kialakításához a reakcióelegyet szobahõfokon 6 órán át kevertem.
Ezalatt,
egy újabb Schlenk edénybe bemértem 40 mmol jódot (
A 6 órás keverést követõen a reakcióelegyet aceton és cseppfolyós nitrogén elegyével -78°C-ra hûtöttem. A elõzõekben elkészített jód oldatot a csepegtetõtölcsér segítségével hozzáadtam a -78°C-os reakcióelegyhez, majd további fél órán át ezen a hõfokon kevertem. A hûtõfürdõt eltávolítva a reakcióelegyet keverés közben szobahõmérsékletûre melegedni.
A reakcióelegyet a jód-ferrocén elõállításánál elvégzett feldolgozási eljárással megegyezõ módon dolgoztam fel.
A tiszta 1,1'-dijód-ferrocént a maradék ferrocéntõl és a melléktermékként keletkezõ jód-ferrocéntõl oszlopkromatográfiával választottam el. Állófázisként Brockmann-II, neutrális alumínium-oxidot használtam, a kromatográfia eluense n-hexán volt.
NaCl-os jeges hûtés közben -5°C-on egy 100 cm3-es gömblombikba 30 ml metanolhoz cseppenként hozzáadtam 0,11 mol tionil-kloridot. 10 perc keverés után spatulányi adagokban hozzáadtam 0,1 mol aminosavat, majd újabb 15 percig kevertem. Ezután a sólé fürdõt vízfürdõre cseréltem és 40°C-on kb. 2 órán át kevertem. Ezt követõen a metanolt ledesztilláltam a reakcióelegyrõl. A lepárolt reakcióelegyet vízben oldottam és NH4OH oldattal semlegesítettem, majd a vizes oldatot szilárd nátrium-kloriddal telítettem. A NaCl-al telített vizes oldatból többszöri izopropil-alkoholos extrakcióval nyertem ki az aminosav-metilészter-hidrokloridot.
Egy saválló autoklávba inert körülmények között bemértem 0,5 mmol jód-ferrocént, 0,025 mmol Pd(OAc)2-t, 0,05 mmol PPh3-t, 2,5 mmol aminosav-észter-hidrokloridot, a táblázatokban megadott mennyiségû bázist és 12,5 ml vízmentes toluolt vagy THF-et. Ezután az autokláv reakcióterét a kívánt szén-monoxid nyomás alá helyeztem és a megfelelõ hõmérsékletû olajfürdõben mágneses keverõ segítségével kevertem. A reakció elõrehaladtát vékonyrétegkromatográfiás, illetve gázkromatográfiás módszerekkel követtem.
A reakcióelegybõl leszûrtem a kivált fém palládiumot, majd vákuumdesztilláció segítségével eltávolítottam az oldószert. A desztillációs maradékot 20 ml dietil-éterben feloldottam, ezt az oldatot 2x20 ml 5%-os H3PO4 oldattal, majd 20 ml telített NaHCO3 oldattal, végül 20 ml telített NaCl oldattal mostam. Az extrakciós lépések után az éteres fázist vízmentes Na2SO4-tal szárítottam, majd a szárítószert kiszûrtem és az étert desztillációval eltávolítottam. Feldolgozás után a terméket oszlopkromatográfiával tisztítottam. Állófázisként Al2O3-ot, eluensként toluol/EtOAc 2/1 arányú elegyét használjuk. A 6a és 6b termékeket szintén Al2O3 oszlopon, EtOAc/MeOH 10/1 eleggyel választottam szét.
Egy saválló autoklávba inert körülmények között bemértem 0,5 mmol 1,1'-dijód-ferrocént, 0,05 mmol Pd(OAc)2-t, 0,1 mmol PPh3-t, megfelelõ mennyiségû primer és/vagy szekunder amint/aminokat, bázisként hozzáadtam 500 ml trietil-amint és oldószerként 25 ml vízmentes toluolt használtam. Ezután az autokláv reakcióterét a kívánt szén-monoxid nyomás alá helyeztem és a megfelelõ hõmérsékletû olajfürdõben mágneses keverõ segítségével kevertem. A reakció elõrehaladtát vékonyrétegkromatográfiás, illetve gázkromatográfiás módszerekkel követtem.
A reakció befejeztével a reakcióelegyet mostam 5%-os H3PO4 oldattal, telített NaHCO3 oldattal, végül telített NaCl oldattal. A szerves fázist Na2SO4-al szárítottam, az oldószert vákuumban ledesztilláltam. A keletkezõ termékeket oszlopkromatográfiával különítettem el egymástól, illetve a kísérõ szennyezésektõl. Állófázisként Brockmann-II, neutrális alumínium-oxidot használtam, az eluens toluol : etil-acetát = 1:1-5 arányú oldószerelegy volt.
Egy Schlenk edénybe inert körülmények között bemértem 0,5 mmol 1'-jód-ferrocénkarboxamidot, vagy -glioxamidot, 0,025 mmol Pd(PPh3)4-t, 0,55 mmol vinil-tributil-sztannánt és oldószerként 7,5 ml toluolt használtam. A reakcióelegyet 100°C-os olajfürdõben mágneses keverõ segítségével kevertem. A reakció elõrehaladását gázkromatográfiás módszerrel követtem.
A reakció végeztével az elegyet kálium-fluorid vizes oldatával kiráztam és a szerves fázist elkülönítettem, majd Na2SO4-al szárítottam. A toluolt vákuumdesztillációval eltávolítottam, és a terméket metanolból átkristályosítottam.
Bemértem 0,5 mmol 1'-jód-N,N-dietil-ferrocénkarboxamidot, 0,025 mmol Pd(OAc)2-ot, 0,05 mmol PPh3-t, 0,75 mmol nátrium-tetrafenil-borátot egy szeptumos feltéttel, gázbevezetõvel, gumiballonnal és golyós hûtõvel ellátott háromnyakú lombikba és a rendszert szén-monoxid atmoszféra alá helyeztem. Majd a reakcióelegyhez hozzámértem 500 ml trietil-amint és 7,5 ml toluolt. Az elegyet 100°C-on 6 órán át kevertem.
A reakció befejeztével a reakcióelegyet mostam 5%-os H3PO4 oldattal, telített NaHCO3 oldattal, végül telített NaCl oldattal. A szerves fázist Na2SO4-al szárítottam, az oldószert vákuumban ledesztilláltam. A tiszta 1'-benzoil-N,N-dietil-ferrocénkarboxamidot oszlopkromatográfiával különítettem el a szennyezésektõl. Állófázisként Brockmann-II, neutrális alumínium-oxidot használtam, eluensként 3:1 arányú benzol: etil-acetát oldószerelegyet alkalmaztam.
Egy saválló autoklávba inert körülmények között bemértem 0,5 mmol 1,1'-dijód-ferrocént, 0,05 mmol Pd(OAc)2-t, 0,1 mmol PPh3-t, a kétféle aminosav-metil-észter-hidrokloridból összesen 1 mmol-nyit, valamit 1 mmol cézium-karbonátot és 25 ml vízmentes toluolt. Ezután az autokláv reakcióterét a kívánt 40 bar szén-monoxid nyomás alá helyeztem és a megfelelõ hõmérsékletû olajfürdõben mágneses keverõ segítségével kevertem. A reakció elõrehaladtát vékonyrétegkromatográfiás, illetve gázkromatográfiás módszerekkel követtem.
A reakció befejeztével a reakcióelegyet mostam 5%-os H3PO4 oldattal, telített NaHCO3 oldattal, végül telített NaCl oldattal. A szerves fázist Na2SO4-al szárítottam, az oldószert vákuumban ledesztilláltam. A keletkezõ termékeket oszlopkromatográfiával különítettem el egymástól, illetve a kísérõ szennyezésektõl. Állófázisként Brockmann-II, neutrális alumínium-oxidot használtam, az eluens összetétele toluol : etil-acetát = 1:1-5 arányú oldószerelegy volt.
Egy 100 cm3-es gömblombikba inert körülmények között bemértem 7,5 ml diklór-metánt, hozzáadtam 1 mmol N-ferrocenoil-glicin-metil-észtert és 1,5 mmol alumínium-kloridot. A reakcióelegyet -30°C-ra hûtöttem és ezen a hõfokon cseppenként hozzáadtam 1 mmol acetil-klorid 7,5 ml diklór-metánnal inert körülmények között készített oldatát. A reakcióelegyet -30°C-on 1 órán át kevertem, majd hagytam 0°C-ra melegedni és ezen a hõfokon további 30 percen keresztül kevertem.
A reakció végén a feleslegben maradt alumínium-kloridot víz hozzáadásával elbontottam, majd többszöri vizes mosással semlegesre mostam. A szerves fázist Na2SO4-al szárítottam, az oldószert lepároltam és tisztán megkaptam az 1'-acetil-N-ferrocenoil-glicin-metil-észter terméket.
A reakciók során a
ferrocénvázas kiindulási anyagok átalakulásának mértékét, valamint a reakciók
termékösszetételét gázkromatográfiás úton határoztam meg. A GC vizsgálatok
Hewlett-Packard típusú készüléken történtek eikozan belsõ standard
alkalmazásával, HP-5890 típusú,
A tiszta termékek szerkezetét 1H- és 13C-NMR segítségével azonosítottam. A spektrumok CDCl3 oldószerben,TMS belsõ standarddal, Bruker Avance 400 spektrométer alkalmazásával 400 és 100,58 MHz-en készültek.
Az infravörös spektrumokat KBr pasztillában AVATAR 330 FT-IR Thermo Nicolet készüléken vettem fel 400-4000 cm-1 tartományban. Az elemanalízisek mérése 1108 Carlo Erba típusú készüléken történt.
2a-c ismert vegyületek és analitikai adataik jól egyeznek az irodalmi értékekkel [89].
N-Ferrocenoil-glicin-metil-észter (2a)
H-NMR (400 MHz, CDCl3) d 6,11 (d, 5,5Hz, 1H, NH); 4,69 (t, 1,7Hz, 2H, 2,5-cp); 4,35 (t, 1,7Hz, 2H, 3,4-cp); 4,24 (s, 5H, nem szubsztituált cp); 4,15 (d, 5,5Hz, 2H, CH2); 3,79 (s, 3H OCH3). IR (cm-1): 3420, 3266; 1746; 1630; 1543.
Kitermelés:
N-Ferrocenoil-L-alanin-metil-észter (2b)
H-NMR (400 MHz, CDCl3) d: 6,40(d, 5,5 Hz, 1H, NH); 4,74 (qui, 5,5 Hz, 1H, CH); 4,72 (brs, 1H, 2-cp); 4,65 (brs, 1H, 5-cp); 4,33 (brs, 2H, 3,4-cp); 4,20 (s, 5H, nem szubsztituált cp); 3,77 (s, 3H, OCH3); 1,47(d, 5,5 Hz, 3H, CH3). IR (cm-1): 3430, 3245; 1730; 1635; 1540.
Kitermelés: 87 %
N-Ferrocenoil-L-fenilalanin-metil-észter (2c)
H-NMR (400 MHz, CDCl3) d 7,24 (m, 5H, Ph); 6,02 (brs, 1H, NH); 5,01 (m, 1H, CH); 4,61 (brs, 1H, 2-cp); 4,57 (brs, 1H, 5-cp); 4,31 (brs, 2H, 3,4-cp); 4,11 (s, 5H, nem szubsztituált cp); 3,75 (s, 3H, OCH3); 3,18 (m, 2H, CHaHb). 13C-NMR (100,5MHz, CDCl3): 172,4 (CO); 170,0(CO); 136,1; 129,2; 128,7; 127,3; 75,4; 70,5; 69,7; 68,3; 68,0; 52,8; 52,3; 38,1. IR (cm-1): 3420, 3240; 1728; 1634; 1540. MS (m/e): 391 (M+)/65; 229/45; 213/100; 185//57; 129/62; 121/55; 91/38; 56/25.
Kitermelés
N-Ferrocenoil-L-metionin-metilészter (2d)
H-NMR (400 MHz, CDCl3) d 6,45 (brs, 1H, NH); 4,85 (m, 1H, CH); 4,71 (brs, 1H, Cp-2); 4,65 (brs, 1H, Cp-5); 4,34 (brs, 2H, Cp-3,4); 4,21 (s, 5H, Cp); 3,77 (s, 3H, OCH3); 2,55 (t, 7Hz, S-CH2); 2,3-1,9 (m, 2H, CH2); 2,12 (s, 3H, S-CH3). IR (KBr, (cm-1)): 3268; 1744; 1625; 1533. MS (m/e): 375 (M+)/69; 213/100; 185/62; 129/37; 121/20; 56/25. Elemanalízis C H FeNO3S (375.27) számított: C, 54.41; H, 5.64; N, 3.73; mért: C, 54.22; H, 5.82; N, 3.61.
Kitermelés: 75%.
N-Ferrocenoil-L-prolin-metil-észter (2e)
H-NMR (400 MHz, CDCl3) d 4,85 (brs, 1H, Cp-2); 4,69 (brs, 1H, Cp-5); 4,61 (brs, 1H, CHCOO); 4,34 (brs, 2H, Cp-3,4); 4,25 (s, 5H, Cp); 3,94 (m, 1H, NCHa); 3,81 (m, 1H, NCHb); 3,76 (s, 3H, OCH3); 2,2 (m, 2H, CH2); 1,98 (m, 2H, CH2). 13C-NMR(100,5MHz, CDCl ):173,0; 169,6; 71,3; 70,5; 70,2; 69,7; 60,1; 52,1; 48,3; 28,7; 25,6. IR (KBr, (cm-1)): 1740; 1590. MS (m/e): 341 (M+)/79; 213/100; 185/45; 129/45; 121/47; 56/25. Elemanalízis C H FeNO3 (341.19) számított: C, 59.85; H, 5.61; N, 4.11; mért: C, 60.02; H, 5.50.; N, 4.23.
Kitermelés: 84 %.
N-Ferrocenil-glioxil-glicin-metil-észter (3a)
H-NMR (400 MHz, CDCl3) d 7,64 (brs, 1H, NH); 5,31 (s, 2H, Cp-2,5); 4,71 (s, 2H, Cp-3,4); 4,24 (s, 5H, Cp); 4,14 (d, 5.6Hz, 2H, CH2); 3,79 (s, 3H, OCH3). 13C-NMR(100,5MHz, CDCl ): 189,7; 169,5; 161,8; 74,5; 74,4; 74,3; 72,2; 70,6; 69,9; 52,5; 40,9 IR (KBr, (cm-1)): 3430; 1740; 1680; 1650; MS (m/e): 329 (M+)/79; 213/100; 185/82; 129/74; 121/47; 56/53. Elemanalízis C H FeNO4 (329.14) számított: C, 54.74; H, 4.59; N, 4.26; mért: C, 54.92; H, 4.71; N, 4.17.
Kitermelés: 89 %.
N-Ferrocenil-glioxil-L-alanin-metil-észter (3b)
H-NMR (400 MHz, CDCl3) d 7,61 (d, 8Hz, 1H, NH); 5,35 (brs; 1H, Cp-2); 5,25 (brs, 1H, Cp-5); 4,70 (brs, 2H, Cp-3,4); 4,63 (quint., 8 Hz , 1H, CH); 4,22 (s, 5H, Cp); 3,78 (s, 3H, OCH3); 1,51 (d, 8 Hz, 3H, -CH-CH3). 13C-NMR(100,5MHz, CDCl IR (KBr, (cm-1)): 3320, 1748; 1683; 1650. MS (m/e): 343 (M+)/66; 213/100; 185/49; 129/41; 121/22; 56/13. Elemanalízis C H FeNO4 (343.16) számított: C, 56.00; H, 4.99; N,4.08; mért: C, 56.17; H, 5.12; N, 4.17.
Kitermelés: 28 %.
N-Ferrocenil-glioxil-L-fenilalanin-metil-észter (3c)
H-NMR (400 MHz, CDCl3) d 7,24 (m, 5H, Ph); 6,50 (brs, 1H, NH); 5,25 (brs, 2H, Cp-2,5); 4,90 (m, 1H, CH); 4,68 (brs, 2H, Cp-3,4); 4,14 (s, 5H, Cp); 3,74 (s, 3H, OCH3); 3,18 (m, 2H, CHaHb). 13C-NMR(100,5MHz, CDCl ): 189,7; 171,2; 161,1; 135,7; 129,2; 128,8; 127,3; 74,4; 74,3; 72,2; 72,1; 70,5; 53,1; 52,5; 38,0. IR (KBr, (cm-1)): 3360; 1736; 1683; 1642. MS (m/e): 419 (M+)/35; 213/71; 185/62; 129/70; 121/45; 91/100; 56/26. Elemanalízis C H FeNO4 (419.26) számított: C, 63.03; H, 5.05; N, 3.34;mért: C, 62.,91; H, 5.14; N,3.22.
Kitermelés: 26 %.
N-Ferrocenil-glioxil-L-metionin-metil-észter (3d)
H-NMR (400 MHz, CDCl3) d 7,73 (d, 8 Hz,1H,NH); 5,34 (s, 1H, Cp-2); 5,26 (s, 1H, Cp-5); 4,76 (m, 1H, CH); 4,71 (brs, 2H, Cp-3,4); 4,22 (s, 5H, Cp); 3,78 (s, 3H, OCH3); 2,57 (t, 8 Hz, S-CH2); 2,29-2,0 (m, 2H, CH2); 2,11 (s, 3H, S-CH3). 13C-NMR(100,5MHz, CDCl ): 189,7; 171,5; 161,4; 74,6; 74,5; 74,4; 72,5; 71,9; 70,6; 52,7; 51,4; 31,5; 30,1; 15,5. IR (KBr, (cm-1)): 3360; 1740; 1683; 1642. MS (m/e): 403 (M+)/85; 213/100; 185/53; 129/48; 121/24; 56/13. Elemanalízis C H FeNO3S (403.28) számított: C, 53.61; H, 5.25; N, 3.47; mért: C, 53.84; H, 5.11; N, 3.59.
Kitermelés: 37 %.
N-Ferrocenil-glioxil-L-prolin-metil-észter (3e)
H-NMR (400 MHz, CDCl3) d 5,01 (brs, 2H, Cp-2,5); 4,73 (brs, 1H, CHCOO); 4,62 (brs, 2H, Cp-3,4); 4,3 (s, 5H, Cp); 3,8 (s, 3H, OCH3); 3,65 (m, 2H, N-CH2); 1,9-2,3 (m, 4H, CH2-CH2). 13C-NMR(100,5MHz, CDCl ): 189,2; 172,1; 164,5; 74,9 ; 73,8; 73,7; 70,6; 70,5; 70,4; 58,5; 52,4; 47,6; 29,0; 25,0. IR (KBr, (cm-1)): 1744; 1665; 1634. MS (m/e): 369 (M+)/43; 213/100; 185/49; 129/39; 121/26; 56/22. Elemanalízis C H FeNO4(369.20) számított: C, 58.56; H, 5.19; N, 3.79; mért: C, 58.41; H, 5.23; N, 3.87.
Kitermelés: 30 %.
1-Ferrocenoil-1,8-diaza-cikloundekan-2-on (6a)
H-NMR (400 MHz, CDCl3) d 7,38 (brs, 1H, NH); 4,80 (brs; 2H, Cp-2,5); 4,29 (brs, 2H, Cp-3,4); 4,19 (s, 5H, Cp); 3,54 (m, 2H, C(O)NCH2); 3,34 (m, 4H, CH2NCH2); 2,59 (m, 2H, C(O)CH2); 1,72 (m, 8H, 4CH2). C-NMR(100,5MHz, CDCl ): 177,3; 170,2; 70,3; 70,2; 69,7; 68,3; 49,5; 44,7; 37,2; 34,7; 30,0; 28,4; 27,3; 23,5. IR (KBr, (cm-1)): 1650; 1638. MS: 382 (M+). Elemanalízis C H FeN2O2 (382.29) számított: C, 62.84; H, 6.86; N, 7.33; mért: C, 62.97; H, 6.71; N, 7.41.
1-Ferrocenil-glioxil-1,8-diaza-cikloundekan-2-on (6b)
H-NMR (400 MHz, CDCl3) d 8.00 (brs, 1H, NH); 5.29 (brs; 2H, Cp-2,5); 4.65 (brs, 2H, Cp-3,4); 4.20 (s, 5H, Cp); 3.47 (m, 2H, C(O)NCH2); 3.34 (m, 4H, CH2NCH2); 2.55 (m, 2H, C(O)CH2); 1.68 (m, 8H, 4CH2). 13C-NMR(100,5MHz, CDCl IR (KBr, (cm-1)):. 1700; 1646; 1634. MS: 410 (M+). Elemanalízis C H FeN2O3 (410.30) számított: C, 61.48; H, 6.39; N, 6.83; mért: C,61.32 ; H, 6.51; N, 6.75.
1'-Jód-N,N-dietil-ferrocénkarboxamid (8a)
H-NMR(400 MHz, CDCl d: 4,58 (brs, 2H, Cp-2,5); 4.45 (brs, 2H, Cp-
Kitermelés: 35 %.
1'-Jód-N,N-dibutil-ferrocénkarboxamid (8b)
H-NMR(400 MHz, CDCl d: 4,53 (t, 1,8 Hz, 2H, Cp); 4,41 (t, 1,8 Hz, 2H, Cp); 4,26 (t, 1,8 Hz, 2H, Cp); 4,22 (t, 1,8 Hz, 2H, Cp); 3,37 (t, 7,9Hz, 4H, NCH2); 1,57 (q, 4H, 7,9Hz, NCH2CH2); 1,31 (m, 4H, N(CH2)2CH2); 0,92 (t, 6H, 7,9Hz, N(CH2)3CH3). IR (KBr, (cm-1)): 1610. MS (m/e): 467(M+)/100; 339/92; 311/85; 247/62; 213/123;183/169. Elemanalízis C H FeINO (467,17): C, 48,85; H, 5,61; N, 3,00; számított: C, 48,97, H, 5,47 ; N, 3,12.
Kitermelés: 31 %.
1'-Jód-morfolino-ferrocénkarboxamid (8c)
H-NMR(400 MHz, CDCl ) d: 4,51 (t, 1,6 Hz, 2H, Cp); 4,43 (t, 1,6 Hz, 2H, Cp); 4,35 (t, 1,6 Hz, 2H, Cp);4,25 (t, 1,6 Hz, 2H, Cp); 3,68 (m, 8H, NCH2CH2O). 13C-NMR(100,5MHz, CDCl ) d: 168,8; 80,3; 76,6; 73,1; 72,6; 71,2; 66,9; 40,1. IR (KBr, (cm-1)): 1618. MS(m/e): 425 (M+)/100; 339/15; 311/8; 213/13; 183/13; 56/10. Elemanalízis C H FeINO2 (425,05): C, 42,39; H, 3,79; N, 3,30; számított: C, 42,60, H, 3,87 ; N, 3,19.
Kitermelés: 14 %.
1'-Jód-piperidino-ferrocénkarboxamid (8d)
H-NMR(400 MHz, CDCl ) d: 4,5 (t, 1,6 Hz, 2H, Cp); 4,43 (t, 1,6 Hz, 2H, Cp); 4,26 (t, 1,6 Hz, 2H, Cp);4,25 (t, 1,6 Hz, 2H, Cp); 3,59 (m, 4H, NCH2); 1,57 (m, 6H, NCH2(CH2)3). 13C-NMR(100,5MHz, CDCl ) d: 168,3; 81,5; 76,4; 73,0; 72,4; 71,2; 46,2; 43,1; 40,1; 29,7; 26,1; 24,8. IR (KBr, (cm-1)): 1616. MS (m/e): 423(M+)/100; 339/11; 311/11, 296/35; 247/11; 213/45; 183/35; 56/15. Elemanalízis C H FeINO (423,08): C, 45,42; H, 4,29; N, 3,31; számított: C, 45,27, H, 4,12 ; N, 3,45.
Kitermelés: 19 %.
1'-Jód-N,N-dietil-ferrocénglioxamid (9a)
H-NMR(400 MHz, CDCl ) d: 4,82 (brs, 2H, Cp); 4,58 (brs, 2H, Cp); 4,53 (brs, 2H, Cp); 4,38 (brs, 2H, Cp); 3,46 (q, 6,8 Hz, 2H, NCH2CH3); 3,25 (q, 6,8 Hz, 2H, NCH2CH3); 1,23 (t, 6,8 Hz, 3H, NCH2CH3); 1,14 (t, 6,8 Hz, 3H, NCH2CH3). 13C-NMR(100,5MHz, CDCl ) d: 197,7; 166,3; 77,5; 76,4; 76,1; 72,4; 72,3; 42,3; 39,9; 39,0; 14,2; 12,8. IR (KBr, (cm-1)): 1642, 1661. MS(m/e): 439 (M+)/100; 339/83; 311/39; 275/15; 183/27; 128/18; 100/43; 72/68; 56/32. Elemanalízis C H FeINO2 (439,08): C, 43,77; H, 4,13; N, 3,19; számított: C, 43,52, H, 4,01; N, 3,31.
Kitermelés: 11 %.
1'-Jód-N,N-dietil-ferrocénglioxamid (9b)
MS(m/e): 495 (M+)/100; 368/13; 340/17; 276/14; 241/30; 213/17; 207/17; 185/11; 152/9; 128/37; 121/33.
1'-Jód-morfolino-ferrocénglioxamid (9c)
H-NMR(400 MHz, CDCl ) d: 4,85 (t, 1,5 Hz, 2H, Cp); 4,60 (t, 1,5 Hz, 2H, Cp); 4,52 (t, 1,5 Hz, 2H, Cp); 4,35 (t, 1,5 Hz, 2H, Cp); 3,69 (m, 6H, morpholine); 3,45 (m, 2H,). 13C-NMR(100,5MHz, CDCl ) d: 196,4; 165,0; 76,7; 76,1; 72,6; 72,1; 70,7; 66,8; 66,7; 46,5; 41,7; 39,8. IR (KBr, (cm-1)): 1646; 1667. MS(m/e): 453 (M+)/91; 339/100; 311/40; 183/21; 128/15; 56/12. Analysis calculated for C H FeINO3 (453,06): C, 42,42; H, 3,56; N, 3,09; Found: C, 42,55, H, 3,67 ; N, 3,21.
Kitermelés: 40 %.
1'-Jód-piperidino-ferrocénglioxamid (9d)
H-NMR(400 MHz, CDCl ) d: 4,82 (brs, 2H, Cp); 4,55 (brs, 2H, Cp); 4,50 (brs, 2H, Cp); 4,35 (brs, 2H, Cp); 3,59 (m, 2H, NCH2); 3,30 (m, 2H, NCH2); 1,62 (m, 4H, NCH2CH2); 1,54 (m, 2H, N(CH2)2CH2). IR (KBr, (cm-1)): 1644; 1662. MS(m/e): 451(M+)/22; 339/62; 311/32; 183/48; 128/54; 121/39; 56/100. Elemanalízis C H FeINO2 (451,09): C, 45,27; H, 4,02; N, 3,11; számított: C, 45,45; H, 4,15; N, 3,21.
Kitermelés: 38 %.
1,1'-N,N-dietil-ferrocéndikarboxamid (10a)
Spektroszkópiai adatai megfelelnek az irodalmi értékeknek. [89]
H-NMR(400 MHz, CDCl ) d 4,63 (s, 4H, 2,5,2',5'-fc); 4,36 (s, 4H 3,4,3',4'-fc); 3,50 (m, 8H, N-CH2-CH3); 1,82 (m, 12H, N-CH2-CH3); 13C-NMR (100,5MHz, CDCl3): d 168,96 CO; 80,63; 71,69; 71,35; 42,66; 40,71; 14,57; 12,84; MS (m/e): 384 (M+)/100; 312/5; 285/11; 241/18; 220/11; 213/11; 121/33.
Kitermelés
1,1'-N,N-dibutil-ferrocéndikarboxamid (10b)
H-NMR(400 MHz, CDCl ) d: 4,59 (t, 1,9 Hz, 4H, Cp); 4,34 (t, 1,9 Hz, 4H, Cp); 3,35 (t, 7,8Hz, 8H, NCH2); 1,58 (m, 8H, NCH2CH2); 1,15 (m, 8H, N(CH2)2CH2); 0,89 (m, 12H, N(CH2)3CH3). IR (KBr, (cm-1)): 1618. MS(m/e): 496 (M+)/100; 368/12; 340/17; 241/32; 128/35; 121/32; 57/38; 56/12.
Kitermelés: 68 %.
1,1'-Morfolino-ferrocéndikarboxamid (10c)
Spektroszkópiai adatai megfelelnek az irodalmi értékeknek. [90]
H-NMR(400MHz, CDCl3) d: 4,57 (brs, 4H); 4,37 (brs, 4H); 3,66 (m, 12H); 3,33 (m, 4H). IR (KBr, (cm-1)): 1616. MS (m/e): 412 (M+)/100; 327/12; 299/20; 241/57; 234/32; 213/21; 207/21; 185/18; 149/19; 129/21; 121/95.
Kitermelés: 72%
1,1'-Piperidino-ferrocéndikarboxamid (10d)
Spektroszkópiai adatai megfelelnek az irodalmi értékeknek. [90]
H-NMR(400MHz, CDCl3) d: 4,59 (brs, 4H); 4,36 (brs, 4H); 3,58 (m, 8H); 1,58 (m, 12H). IR (KBr, (cm-1)): 1618. MS (m/e): 408 (M+)/52; 325/12; 297/24; 241/16; 213/12; 204/24; 186/21; 140/18; 121/46; 84/100.
Kitermelés: 75%.
1,1'-N-butil-ferrocéndikarboxamid (10e)
H NMR(400MHz, CDCl3) d: 6,67 (brs, 2H, NH); 4,46 (s, 4H); 4,3 (s, 4H); 3,36 (q, 6,8 Hz, 4H); 1,61 (m, 4H); 1,41 (m, 4H); 0,98 (t, 7,2 Hz, 6H). IR (KBr, (cm-1)): 3321; 1630 ; 1552. MS (m/e): 384(M+)/52; 312/14; 285/15; 241/28; 190/28; 146/28; 121/100.
Kitermelés: 68%.
1'-(N',N'-dietil-karbamoil)-N,N-dibutil-ferrocénkarboxamid (10ab)
H-NMR(400MHz, CDCl ) d: 4,62 (brs, 2H, Cp); 4,58 (brs, 2H, Cp); 4,35 (brs, 2H, Cp); 4,27 (brs, 2H Cp); 3,44 (q, 6,4 Hz, 4H, NCH2CH3); 3,35 (t, 8Hz, 4H, NCH2CH2); 1,54 (m, 4H, NCH2CH2); 1,26 (m, 4H, N(CH2)2CH2); 1,17 (t, 6,4 Hz, 6H, NCH2CH3); 0,91 (t, 8Hz, 6H, N(CH2)3CH3). 13C-NMR(100,5MHz, CDCl ) d: 169,2; 169,0; 81,1; 80,6; 71,7; 71,6; 71,5; 71,3; 48,5; 46,4; 42,5; 40,9; 31,4; 29,6; 20,2; 20,1; 13,8. IR (KBr, (cm-1)): 1614. MS(m/e): 440 (M+)/100; 241/37; 220/43; 128/65; 121/80; 70/63; 57/35, 56/32. Elemanalízis C H FeN2O2 (440,41): C, 65,45; H, 8,24; N, 6,36; számított: C, 65,63, H, 8,31 ; N, 6,28.
Kitermelés: 77 %.
1'-(Morfolino-karbamoil)-N,N-dietil-ferrocénkarboxamid (10ac)
H-NMR (400MHz, CDCl3) d: 4,62 (s, 2H, 2,5-Cp); 4,58 (s, 2H,
Kitermelés: 85%.
1'-(Piperidino-karbamoil)-N,N-dietil-ferrocénkarboxamid (10ad)
H-NMR (400MHz, CDCl3) d: 4,63 (s, 2H, 2,5-Cp); 4,57 (s, 2H,
Kitermelés:58%.
1'-(N'-butil-karbamoil)-N,N-dietil-ferrocénkarboxamid (10ae)
H NMR(400MHz, CDCl3) d: 8,42 (brs, 1H); 4,60 (s, 2H); 4,36 (brs, 4H); 4,32 (s, 2H); 3,47 (m, 2H); 3,38 (m, 4H); 1,29-1,65 (m, 4H); 1,19 (m, 6H); 0,95 (t, 7,5Hz, 3H). IR (KBr, (cm-1)): 3315; 1646, 1613; 1535. MS (m/e): 384 (M+)/38; 313/8; 285/17; 241/26; 229/12; 220/22; 213/22; 190/20; 163/20; 149/18; 128/38; 121/86; 72/100; 56/68. Elemanalízis C20H28FeN2O2 (384,30): C, 62,51; H, 7,34; N, 7,29; számított: C, 62,15; H, 7,61; N, 7,62.
Kitermelés: 44%.
1'-(N'-butil-karbamoil)-N,N-dibutil-ferrocénkarboxamid (10be)
H NMR(400MHz, CDCl3) d: 8,53 (brs, 1H); 4,59 (s, 2H); 4,28 (brs, 6H); 3,10-3,42 (m, 6H); 1,10-1,65 (m, 12H); 0,88-1,00 (m, 9H). IR (KBr, (cm-1)): 3297; 1650; 1610; 1535. MS (m/e): 440(M+)/100; 312/18; 241/44; 128/56; 121/63; 56/19. Elemanalízis C24H36FeN2O2 (440,41): C, 65,45; H, 8,24; N, 6,36; számított: C, 65,81; H, 8,56; N, 6,12.
Kitermelés: 36%.
1'-(Piperidino-karbamoil)-morfolino-ferrocénkarboxamid (10cd)
H NMR(400MHz, CDCl ) d: 4,59 (s, 2H, Cp); 4,56 (s, 2H, Cp); 4,39 (s, 2H); 4,36 (s, 2H Cp); 3,67 (m, 8H); 3,56 (m, 4H); 1,55 (m, 6H). IR (KBr, (cm-1)): 1615. MS (m/e): 410(M+)/100; 297/19; 241/46; 207/52; 186/35; 128/28.
Kitermelés: 51%.
1'-(Morfolino-karbamoil)-N-butil-ferrocénkarboxamid (10ce)
H NMR(400MHz, CDCl3) d: 8,32 (brs, 1H); 4,57 (s, 2H); 4,33 (brs, 4H); 4,31 (s, 2H); 3,64 (m, 8H); 3,36 (q, 7.5Hz, 2H); 1,60 (qui, 7,5Hz, 2H); 1,42 (m, 2H); 0,95 (t, 7,5Hz, 3H). IR (KBr, cm-1): 3252; 1642; 1593; 1552. MS (m/e): 398 (M+)/34; 313/8; 285/14; 241/34; 213/14; 185/13; 149/20; 121/100; 56/71. Elemanalízis C20H26FeN2O3 (398,28): C, 60,31; H, 6,58; N, 7,03; számított: C, 60,78; H, 6,79; N, 6,75.
Kitermelés: 62%.
1'-(N',N'-dietil-karbamoil)-N,N-dietil-ferrocénglioxamid (11a)
MS (m/e): 412 (M+)/100; 312/30; 284/6; 241/74; 207/56; 162/9; 121/33.
1'-vinil-N,N-dietil-ferrocénkarboxamid (13a)
H-NMR(400MHz, CDCl d: 6,42 (dd, 10,8 Hz, 17,6Hz, 1H, CH=CH2); 5,35 (dd, 1,2 Hz, 17,6 Hz, 1H, CH=CHAHB); 5,05 (dd, 1,2 Hz, 10,8 Hz, 1H, CH=CHAHB); 4,53 (t, 2 Hz, 2H, Cp); 4,37 (t, 2 Hz, 2H, Cp); 4,26 (t, 2 Hz, 2H, Cp); 4,22 (t, 2 Hz, 2H, Cp); 3,49 (q, 6,8 Hz, 4H, NCH2CH3); 1,20 (t, 6,8Hz, 6H, NCH2CH3). C-NMR(100,5MHz, CDCl d: 169,2; 133,8; 112,1; 84,7; 79,5; 71,4; 70,7; 70,6; 68,4; 42,6; 40,4; 14,3; 12,8. IR (KBr, (cm-1)): 1608. MS(m/e): 311 (M+)/100; 239/56; 212/51; 210/50; 121/55; 56/34. Elemanalízis C H FeNO (311,21): C, 65,61; H, 6,80; N, 4,50; számított: C, 65,78, H, 6,71 ; N, 4,57.
Kitermelés: 69 %.
1'-Vinil-morfolino-ferrocénglioxamid (14c)
H-NMR(400MHz, CDCl d: 6,34 (dd, 10,8 Hz, 17,6Hz, 1H, CH=CH2); 5,36 (dd, 1,2 Hz, 17,6 Hz, 1H, CH=CHAHB); 5,11 (dd, 1,2 Hz, 10,8 Hz, 1H, CH=CHAHB); 4,76 (brs, 2H, Cp); 4,55 (brs, 2H, Cp); 4,46 (brs, 2H, Cp); 4,38 (brs, 2H, Cp); 3,74 (m, 2H,); 3,69 (m, 2H,); 3,64 (m, 2H,); 3,42 (m, 2H,). C-NMR(100,5MHz, CDCl d: 196,9; 165,2; 132,6; 113,4; 86,0; 75,5; 75,2; 71,6; 71,2; 68,9; 66,8; 66,7; 46,4; 41,7. IR (KBr, (cm-1)): 1630; 1652. MS(m/e): 353 (M+)/36; 239/100; 211/82; 121/57; 56/55. Elemanalízis C H FeNO3 (353,20): C, 61,21; H, 5,42; N, 3,97; számított: C, 61,37, H, 5,31 ; N, 3,85.
Kitermelés: 85 %.
1'-Vinil-piperidino-ferrocénglioxamid (14d)
H-NMR(400MHz, CDCl d: 6,36 (dd, 10,4 Hz, 17,6Hz, 1H, CH=CH2); 5,36 (dd, 1,2 Hz, 17,6 Hz, 1H, CH=CHAHB); 5,11 (dd, 1,2 Hz, 10,4 Hz, 1H, CH=CHAHB); 4,74 (t, 2 Hz, 2H, Cp); 4,52 (t, 2 Hz, 2H, Cp); 4,47 (t, 2 Hz, 2H, Cp); 4,39 (t, 2 Hz, 2H, Cp); 3,6 (m, 2H, NCH2); 3,31 (m, 2H, NCH2); 1,62 (m, 4H, NCH2CH2); 1,55 (m, 2H, N(CH2)2CH2). C-NMR(100,5MHz, CDCl d: 198,0; 165,3; 132,8; 113,2; 85,9; 75,7; 74,9; 71,7; 71,1; 68,9; 47,1; 42,2; 26,2; 25,5; 24,5. IR (KBr, (cm-1)): 1630 ; 1650. MS(m/e): 351 (M+)/56; 239/100; 211/52; 121/32; 56/27. Elemanalízis C H FeNO2 (351,23): C, 64,97; H, 6,03; N, 3,99; számított: C, 65,11, H, 6,11 ; N, 4,10.
Kitermelés: 82 %.
1'-benzoil-N,N-dietil-ferrocénkarboxamid (15a)
H-NMR(400MHz, CDCl d: 7,88 (d, 6Hz, 2H, Ph); 7,52 (t, 6Hz, 1H, Ph); 7,44 (t, 6Hz, 2H, Ph); 4,93 (brs, 2H, Cp); 4,65 (brs, 2H, Cp); 4,59 (brs, 2H, Cp); 4,29 (brs, 2H, Cp); 3,37 (q, 7,2 Hz, 4H, NCH2CH3); 1,12 (t, 7,2 Hz, 6H, NCH2CH3). C-NMR(100,5MHz, CDCl d: 198,6; 168,2; 139,5; 131,7; 128,2; 127,5; 81,6; 79,1; 74,9; 73,0; 71,8; 71,5; 42,5; 40,7; 14,3; 12,8. IR (KBr, (cm-1)):1610; 1632. MS(m/e.): 389 (M+)/100; 317/30; 290/27; 260/31; 133/38; 72/28; 56/62. Elemanalízis C H FeNO2 (389,28): C, 67,88; H, 5,96; N, 3,60; számított: C, 67,95, H, 5,85 ; N, 3,72.
Kitermelés: 78 %.
N,N-Dietil-ferrocénkarboxamid (16a)
A korábban már karakterizált vegyülettel [91] való azonosságát GC-MS méréssel igazoltam.
MS (m/e): 285(M+)/100; 213/44; 186/44; 185/31; 121/40; 129/35; 56/32.
Morfolino-ferrocénkarboxamid (16c)
A korábban már karakterizált vegyülettel [90] való azonosságát GC-MS méréssel igazoltam.
MS (m/e): 299(M+)/60; 213/38; 185/38; 129/50; 56/100.
17a-c [28] 17d [92] és 17bc [28] ismert vegyületek és analitikai adataik (1H NMR és/vagy MS) jól egyeznek az irodalmi értékekkel.
Fe[C5H4-CO-Gly-OMe]2 (17a)
H NMR (400MHz CDCl3) 7,97 (brs, 2H, NH); 4,84 (brs, 4H, Cp,); 4,51 (brs, 4H, Cp); 4,32 (brs, 4H, CH2); 3,84 (s, 6H, OCH3).
Fe[C5H4-CO-Ala-OMe]2 (17b)
H NMR (400 MHz, CDCl3) 7,73 (d, 1H, NH); 4,89 (brs, 2H, Cp,); 4,84 (m, 2H, CH); 4,74 (brs, 2H, Cp); 4,54 (brs, 2H, Cp); 4,33 (brs, 2H, Cp); 3,80 (s, 6H, OCH3); 1,38 (d, 7,5 Hz, 6H, CH-CH3). MS (m/e): 444 (M+)/100; 412/2; 384/5; 342/9; 271/14; 250/3; 223/3; 190/3; 160/7; 130/16.
Fe[C5H4-CO-Phe-OMe]2 (17c)
MS (m/e): 596 (M+)/100; 434/8; 271/26; 91/10.
Fe[C5H4-CO-Met-OMe]2 (17d)
MS (m/e): 564 (M+)/100; 490/7; 458/3; 433/7; 402/11; 355/13; 310/16; 271/42; 218/34; 190/53; 160/37.
Fe[C5H4-CO-Phe-OMe][C5H4-CO-Gly-OMe] (17ac)
H NMR
(400 MHz, CDCl3) d: 7,87
(brs, 1H, NH); 7,54 (d, 8Hz, 1H, NH); 7,14-7,27 (m, 5H, Ph); 4,95 (m, 1H, CH);
4,85 (brs, 2H, Cp); 4,56 (brs, 2H, Cp); 4,53 (m, 1H, CHa,b-CO2Me); 4,52 (brs, 1H, Cp); 4,50
(brs, 1H, Cp); 4,32 (brs, 2H, Cp); 3,83 (s, 3H, OCH3); 3,79 (s, 3H,
OCH3); 3,69 (dd, 7,6 Hz, 18Hz, 1H, CHa,b-CO2Me);
3,12 (dd, 14 Hz, 4,9Hz, 1H, CHa,b-Ph);
2,84 (dd, 14 Hz, 10,3 Hz, 1H, CHa,b-Ph).
Kitermelés: 47%.
Fe[C5H4-CO-Gly-OMe][C5H4-CO-Met-OMe] (17ad)
H NMR
(400 MHz, CDCl3) d: 7,95
(brs, 1H, NH); 7,72 (brs, 1H, NH); 4,95 (brs, 2H, Cp); 4,88 (brs, 2H, Cp); 4,76
(brs, 2H, Cp); 4,64 (m, 1H, CH); 4,39 (brs, 2H, Cp); 3,85 (s, 3H, OCH3);
3,84 (s, 3H, OCH3); 3,78 (m, 2H, CH2-CO2Me); 2,55 (m, 2H, CH2-S); 2,18
(m, 2H, CH2-CH2S);
2,05 (s, 3H, S-CH3).
Kitermelés: 39%.
Fe[C5H4-CO-Phe-OMe][C5H4-CO-Ala-OMe] (17bc)
H NMR (400 MHz, CDCl3) d: 7,84 (brs, 1H, NH); 7,74 (brs, 1H, NH); 7,30-7,20 (m, 5H, Ph); 5,02 (m, 1H, CH); 4,82-4,94 (m, 3H, Cp+CH); 4,76 (brs, 1H, Cp); 4,71 (brs, 1H, Cp); 4,54 (brs, 2H, Cp); 4,35 (brs, 2H, Cp); 3,87 (s, 3H, OCH3); 3,84 (s, 3H, OCH3); 3,15 (m, 1H, CHa,b-Ph); 2,91 (m, 1H, CHa,b-Ph); 1,42 (d, 7,5 Hz, 6H, CH-CH3). Elemanalízis C26H28N2O6Fe: C, 60,01; H, 5,42; N, 5,38; számított: C, 60,31; H, 5,21; N, 5,49.
Kitermelés: 27%.
Fe[C5H4-CO-Ala-OMe][C5H4-CO-Met-OMe] (17bd)
H NMR
(400 MHz, CDCl3) d: 7,78
(d, 1H, NH); 7,72 (d, 1H, NH); 4,88 (m, 4H, Cp, 2CH); 4,73 (brs, 2H, Cp); 4,54
(brs, 2H, Cp); 4,34 (brs, 2H, Cp); 3,81 (s, 6H, OCH3); 2,66 (m, 1H,
CHa,b-S); 2,54
(m, 1H, CHa,b-S); 2,08 (s, 3H, S-CH3);
2,06 (m, 2H, CH2-CH2S);
1,39 (d, 3H, CH-CH3).
Kitermelés: 40%.
Fe[C5H4-CO-Phe-OMe][C5H4-CO-Met-OMe] (17cd)
H NMR (400 MHz, CDCl3) d: 7,69 (d, 1H, NH); 7,48 (s, 1H, NH); 5,03 (m, 1H, CH-CH2Ph,); 4,90 (m, 2H, Cp); 4,74 (m, 3H, Cp+CH-CH2S); 4,51 (m, 2H, Cp); 4,31 (m, 2H, Cp); 3,81 (s, 6H, OCH3); 3,20 (m, 1H, CHa,b-Ph); 2,90 (m, 1H, CHa,b-Ph); 2,71 (m, 1H, CHa,b-S); 2,55 (m, 1H, CHa,b-S); 2,08 (s, 3H, S-CH3); 2,06 (m, 2H, CH2-CH2S). 13C NMR (100,5 MHz, CDCl3) d: 175,7 (COészter); 170,6 (COamid); 75,9; 72,2; 71,6; 70,5; 70,4; 54,0; 53,1; 51,6; 37,2; 30,9; 15,7. IR (KBr (cm-1)): 3303
Kitermelés: 52%.
1'-Acetil-N-ferrocenoil-glicin-metil-észter (18)
H NMR (400 MHz, CDCl3) d: 6,54 (brs, 1H, NH); 4,88 (s, 2H); 4,65 (s, 2H); 4,55 (s, 2H); 4,40 (s, 2H); 4,16 (s, 2H); 3,78 (s, 3H); 2,38 (s, 3H).
Kitermelés: 99%.
A ferrocén felfedezését követõen számos kutatócsoport irányította figyelmét az addig ismeretlen molekula szerkezetének megismerésére, funkcionalizálására, ami új lendületet adott a fémorganikus kémia fejlõdésének. Hamarosan különbözõ szintézisutak segítségével a legkülönbözõbb ferrocénszármazékok megjelenése színesítette a fémorganikus származékok palettáját. A ferrocénszármazékok kedvezõ tulajdonságaiknak köszönhetõen gyakorlati szempontból mind nagyobb jelentõségre tesznek szert, ami a szintézisük minél gazdaságosabb, hatékonyabb megvalósítását teszik szükségessé. Munkám során N-ferrocenoil-aminosav-észtereket, a ferrocénvázas vegyületek között újszerû ferrocénglioxamidokat és egyéb ferrocénszármazékokat kívántam szintetizálni a ferrocénszármazékok elõállítására eddigiekben nem alkalmazott palládium katalizált homogénkatalitikus karbonilezési eljárással.
A Pannon Egyetem Szerves Kémia Intézeti Tanszékén elkezdett jód-ferrocén karbonilezési kísérletek során új típusú ferrocénglioxamidokat szintetizáltak, ferrocénkarboxamid melléktermék keletkezése mellett.
A munka folytatásaként vizsgáltam a jód-ferrocén palládium katalizált aminokarbonilezését aminosav-észterek mint nukleofil reakciópartnerek jelenlétében. Célom a mono- és dikarbonilezett vegyületek minél szelektívebb elõállítása volt.
Megállapítottam, hogy a reakciók szelektivitását a korábbiakkal ellentétben nem a hõmérséklettel, hanem a megfelelõ bázis alkalmazásával lehet eredményesen befolyásolni. Vizsgálataim eredményeként meghatároztam a ferrocénglioxamidok, illetve ferrocénkarboxamidok elõállításához szükséges optimális reakciókörülményeket. Kimutattam, hogy karbonilezséi körülmények között a bázisként alkalmazott DBU is acilezhetõ.
Az e reakciókban szerzett tapasztalatokat is felhasználva vizsgáltam az 1,1'-dijód-ferrocén aminokarbonilezési reakcióját. A reakciókörülmények megfelelõ megválasztásával 1'-jód-ferrocénkarboxamidokat, illetve -glioxamidokat szintetizáltam, melyeknek további homogénkatalitikus reakcióit is megvalósítottam.
Új, egyszerû és hatékony eljárást dolgoztam ki két különbözõ amidocsoportot tartalmazó 1,1'-ferrocéndikarboxamidok elõállítására, melyekre rendkívül kevés példát találunk az irodalomban.
Ezzel analóg módon nem szimmetrikusan diszubsztituált 1,n'-ferrocenoil-aminosav-észtereket szintetizáltam.
HO-Su: N-Hidroxi-szukcinimid
HO-Bt: 1-Hidroxi-benzotriazol
DCC N,N'-Diciklohexil-karbodiimid
EDC 1-Etil-3-(3-(dimetil-amino)-propil)-karbodiimid-hidroklorid
HBTU: O-Benzotriazol-N,N,N',N'-tetrametil-uronium-hexafluoro-foszfát
TBTU: O-(Benzotriazol-1-il)-N,N,N',N'-tetrametiluronium-tetrafluoroborát
DMAP: 4-(Dimetil-amino)-piridin
CMPI: 2-Klór-1-metil-piridínium-jodid
TFA Trifluor-ecetsav
BOC: Terc-butoxi-karbonil csoport
Kealy, T. J.; Pauson, P. L.; (1951). Nature 168, 1039
Wilkinson, G.; Rosenblum, M.; Whiting, M. C.; Woodward, R. B. J. Am. Chem. Soc 1952, 74, 2125
Fischer, E. O.; Pfab, W. Z. Naturforsch. B 1952, 7, 377
Faigl, F.; Kollár, L.; Kotschy A.; Szepes, L. Szerves fémvegyületek kémiája Nemzeti Tankönyvkiadó 2001, 290
Beer P. D.; Keefe, A. D.; Sikanyika, H. J Chem Soc
Moutet, J.-C.; Saint.Aman, E.; Ungureanu, M.; Visan, T. J. Electroanal. Chem.
Heinze, K.; Schlenker, M. Eur. J. Inorg. Chem. 2005, 66
Alonso, E.; Laband, A.; Raehm, L.; Kern, J.-M.; Astruc, D. C. R. Comptes Rendus de l'Académie des Sciences - Series IIC - Chemistry 1999, 2, 209
Westwood, J.; Coles, S.J.; Collinson, S.R.; Gasser, G.; Green, S.J.; Hursthouse, M.B.; Light, M.E.; Tucker, J.H.R. Organometallics 2004, 23, 946
Collinson, S. R.; Gelbrich, T.; Hursthouse, M. B.; Tucker, J. H. R. Chem. Commun 2001. 555
Ihara, T.;Maruo, Y.; Takenaka, S.;Takagi, M. Nucleic Acids Res. 1996, 24, 4273
Degani, Y.; Heller, A. J. Am. Chem. Soc. 1988, 110, 2615
Weliky, N.; Gould, E. S. J. Am. Chem. Soc. 1957, 79, 2742.
Rausch, M.; Shaw, P.; Mayo, D.; Lovelace, A. M. J. Org. Chem. 1958, 23, 505.
Pberhoff, M.; Duda, L.; Karl, J.; Mohr, R.; Erker, g.; Fröhlich, R.; Grehl, M. Organometallics 1996, 15, 4005
Acton, E. M.; Silverstein, R. M. J. Org. Chem. 1957, 24, 1487.
Galow, T. H.; Rodrigo, J.; Cleary, K.; Cooke, G.; Rotello, V. M. J. Org. Chem. 1999, 64, 3715.
Schetter, B.; Speiser, B. J. Organomet. Chem. 2004, 689, 1472
Seio, K.; Mizuta, M.; Terada, T.; Sekine, M. Tetrahedron Lett. 2004, 45, 6783
Mandal, H. S.; Kraatz, H.-B. J. Organomet. Chem. 2003, 674, 32.
Schlögl, K Monatsh. Chem. 1957, 88, 601.
Herrick, R. S.; Jarret, R. M.; Curran, T. P.; Dragoli, D. R.; Flaherty, M.B.; Lindyberg, S. E.; Slate, R. A.; Thornton, L. C. Tetrahedron Lett. 1996, 37, 5289.
Kraatz, H.-B.; Lusztyk, J.; Enright, G. D. Inorg. Chem. 1997, 36, 2400.
Staveren, van D. R.; Weyhermüller, T.; Metzler-Nolte, N. J. Cem. Soc. Dalton Trans. 2003, 210
Bauer, W.; Polborn, K.; Beck, W. J. Organomet. Chem. 1999, 579, 269.
Kitagawa, K.; Morita, T.; Kawasaki, M.; Kimura, S. J. Polymer. Sci. A 2003, 41, 3493.
Xu, Y.; Kraatz, H.-B. Tetrahedron Lett. 2001, 42, 2601
Kirin, S. I.; Wissenbach, D.; Metzler-Nolte, N. New. J. Chem. 2005, 29, 1168.
Bedford, R. B.; Cazin, C. S. J.; Holder, D. Coord. Chem. Rev. 2004, 248, 2283
Mitchell, T. N. "Organotin Reagents in Cross-coupling", Metal-catalyzed Cross-coupling Reactions (Eds. Diederich, F.; Stang, P. J.) Wiley-VCH, Weinheim, 1998. Chapter 4
Suzuki, A. "Cross-coupling Reactions of Organoboron Compounds with Organic Halides", Metal-catalyzed Cross-coupling Reactions (Eds. Diederich, F.; Stang, P. J.) Wiley-VCH, Weinheim 1998, Chapter 2
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457
Suzuki, A. J. Organomet. Chem. 1999, 576, 147
Kotha, S.; Lahiri, K.; Kashinath, D. Tetrahedron 2002, 58, 9633
Suzuki, A. J. Organomet. Chem. 2002, 653, 83
Negishi, E.; Liu, F. "Palladium- or Nickel-catalyzed Cross-coupling with Organometals Containing Zinc, Magnesium, Aluminum and Zirconium", Metal-catalyzed Cross-coupling Reactions (Eds. Diederich, F.; Stang, P. J.) Wiley-VCH, Weinheim 1998, Chapter 1
Negishi, E.; Hu, Q.; Huang, Z.; Qian, M.; Wang, G. Aldrichimica Acta 2005, 38, 71
Hiyama, T. "Organosilicon Compounds in Cross-Coupling Reactions", Metal-catalyzed Cross-coupling Reactions (Eds. Diederich, F.; Stang, P. J.) Wiley-VCH, Weinheim, 1998, Chapter 10
Spivey, A. C.; Gripton, C. J. G.; Hannah, J. P. Curr. Org. Synth. 2004, 1, 211
Murahashi, S. I. J. Organomet. Chem
Pérez, I.; Sestelo, J. P.; Sarandeses, L. A. J. Am. Chem. Soc
Knapp, R.; Rehahn, M. J. Organomet. Chem. 1993, 452, 235.
Imrie, C; Loubser, C.; Engelbrecht, P.; McCleland, C. W. J. Chem. Soc. 1999, 1, 2513.
Jackson, R. F. W.; Turner, D.; Block, M. H. Synlett 1996, 862.
Sonogashira, K. J. Organomet. Chem. 2002, 653, 46-49.
Böhm, V. P. W.; Hermann, W. A. Eur. J. Org. Chem. 2000, 3679.
Liang, B.; Dai, M.; Chen, J.; Yang, Z. J. Org. Chem. 2005, 70, 391.
Gholap, A. R.; Venkatesan, K.; Pasricha, R.; Daniel, T.; Lahoti, R. J.; Srinivasan, K. V. J. Org. Chem. 2005, 70, 4869.
Yi, C.; Hua, R. J. Org. Chem. 2006, 71, 2535.
Kasahara, A.; Izumi, T.; Maemura, M. Bull. Chem. Soc. Jpn. 1977, 50, 1021.
Lavaste, O.; Plass, J.; Bachmann, P.; Guesmi, S.; Moinet, C.; Dixneuf, P. H. Organometallics 1997, 16, 184.
Yamamoto, T.; Morikita, T.; Maruyama, T.; Kubota, K.; Katada, M. Macromolecules 1997, 30, 5390.
Linder, E.; Ruifa, Z.; Eichele, K.; Ströbele, M. J. Organomet Chem. 2002, 660, 78
Jeffery, T. Tetrahedron Lett. 1985, 26, 2667-2670.
Calo, V.; Nacci, A.; Monopoli, A.; Laera, S.; Cioffi, N. J. Org. Chem. 2003, 68, 2929-2933.
Carlström, A.-S.; Frejd, T. J. Org. Chem. 1990, 55, 4175.
Andersson, C-M.; Hallberg, A.; Jr. Davis, G.D. J. Org. Chem. 1987, 52, 3529
Pedersen, B.; Wagner, G.; Herrmann, R.; Scherer, W.; Meerholz, K.; Schmalzlin, E.; Brauchle, C. J. Organomet. Chem. 1999., 590, 129.
Ozawa, F.; Soyama, H.; Yanaghara, H.; Aoyama, I.; Takino, H.; Izawa, K.; Yamamoto, T.; Yamamoto, A. J. Chem. Soc. 1985, 107, 3235
Lin, Y-S.; Yamamoto, A. Organometallics 1998, 17, 3466.
Colquhoun, H. M.; Thompson, D. J.; Twigg, M. V. Carbonylation Direct Synthesis of Carbonyl Compounds, New York, 1991, 26.
Sakakura, T.; Chaisupakitsin, M.; Hayahi, T.; Tanaka, M. J. Organomet. Chem. 1987, 334, 205
Ozawa, F.; Yamamoto, A. Chem. Lett. 1982, 865.
Kobayashi, T.; Tanaka, M. J. Organomet. Chem. 1982, 233, C64.
Ozawa, F.; Sugimoto, T.; Yuasa, Y.; Santra, M.; Yamamoto, T.; Yamamoto, A. Organometallics 1984, 3, 683.
Ozawa, F.; Sugimoto, T.; Yamamoto, T.; Yamamoto, A. Organometallics 1984, 3, 692
Uozumi, Y.; Arii, T.; Watanabe, T. J. Org. Chem. 2001. 66, 5272.
Tsukada, N.; Ohba, Y.; Inoue, Y. J. Organomet. Chem. 2003. 687, 436.
Skoda-Földes, R.; Szarka, Zs.; Kollár, L.; Dinya, Z.; Horváth, J.; Tuba, Z. J. Org. Chem. 1999, 64,2134.
Szarka, Zs.; Skoda-Földes, R.; Kollár, L.; Horváth, J.; Tuba, Z. Synth. Commun. 2000, 30, 1945.
Skoda-Földes, R.; Székvölgyi, Z.; Kollár, L.; Berente, Z.; Horváth, J.; Tuba, Z. Tetrahedron 2000, 56, 3415.
Szarka, Zs.; Skoda-Földes, R.; Kollár, L. Tetrahedron Lett. 2001, 42, 739.
Watanabe, M.; Araki, S.; Butsugan, Y. J. Org. Chem. 1991, 56, 2218.
Shieh, W-C.; Dell, S.; Repiè, O. J. Org. Chem. 2002, 67, 2188.
Shieh, W-C.; Lozanov, M.; Loo, M.; Repiè, O.; Blacklock, T. J. Tetrahedron Lett. 2003, 44, 4563.
Juneja, T. R.; Garg, D. K.; Schäfer, W. Tetrahedron 1982, 38, 551.
Wang, Y.; Rillema, D. P. Inorg. Chem. Comm. 1998, 1, 27.
Skoda-Földes, R.; Csákai, Z.; Kollár, L.; Horváth, J.; Tuba, Z. Steroids 1995, 60, 812-816
Herrick, R. S.; Jarret, R. M.; Curran, T. P.; Dragoli, D. R.; Flaherty, M. B.; Lyndiberg, S. E.; Slate R. A.; Thornton, l. C. Tetrahedron Lett. 1996, 37, 5289.
Kirin, S. I.; Schatzschneider, U.; de Hatten, X.; Weyhermüller, T.; Metzler-Nolte, N. J. Organomet. Chem. 2006, 691, 3451
Moriuchi, T.; Nomoto, A; Yoshida, K.; Ogawa, A.; Hirao, T. J. Am. Chem. Soc. 2001, 123, 68
Orpen, A.G.; Brammer, L.; Allen, F. H.; Kennard, O.; Watson, D. G.; Taylor, R. J. Chem. Soc., Dalton Trans. 1989, S1-S83
Orpen, A.G.; Brammer, L.; Allen, F. H.; Kennard, O.; Watson, D. G.; Taylor, R. second ed. International Tables for X-ray Crystallography, vol. C Kluwer Academic Publisher, Dordrecht, The Netherlands 1999, 804-888
van Steveren D. R.; Metzler-Nolte, N. Chem. Rev. 2004, 104, 5931
Moriuch, T.; Hirao, T. Chem. Soc. Rev. 2004, 33, 294
Kirin, S. I.; Kraatz, H.-B.; Metzler-Nolte, N. Chem. Soc. Rev. 2006, 35, 348
Herrick, R. S.; Jarret, R. M.; Curran, T. P.; Dragoli, D. R.; Flaherty, M. B.; Lyndiberg, S. E.; Slate R. A.; Thornton, l. C. Tetrahedron Lett. 1996, 37, 5289
Little, F. W.; Eisenthal, R. Organic and Biological Chem. 1960, 82, 1577
Sheehy, M. J.; Gallagher, J. F.; Yamashita, M.; Ida, Y.; White-Colangelo, J.; Johnson, J.; Orlando, R.; Kenny, P. T. M. J. Organomet. Chem. 2004, 689, 151
Hammond, P. J.; Beer, P. D.;
Dudman, C.; Danks,
Szarka, Z.; Skoda-Földes, R.; Kuik, A.; Berente, Z.; Kollár, L. Synthesis 2003, 545
de Hatten, X.; Weyhermüller, T.; Metzler-Nolte, N. J. Organomet. Chem. 2004, 689, 4856.
Találat: 5162